
Abstract
Genome-wide association studies (GWASs) have identified >20 genetic loci associated with human intelligence. However, due to correlations between the trait-associated SNPs, only a few of the loci are confirmed to have a true biological effect. In order to distinguish the SNPs that have a causal effect on human intelligence, we must eliminate the noise from the high degree of linkage disequilibrium that persists throughout the genome. In this study, we apply a novel PAINTOR fine-mapping method, which uses a Bayesian approach to determine the SNPs with the highest probability of causality. This technique incorporates the GWAS summary statistics, linkage disequilibrium structure, and functional annotations to compute the posterior probability of causality for all SNPs in the GWAS-associated regions. We found five SNPs (rs6002620, rs41352752, rs6568547, rs138592330, and rs28371699) with a high probability of causality, three of which have posterior probabilities >0.60. The SNP rs6002620 (NDUFA6), which is involved in mitochondrial function, has the highest likelihood of causality. These findings provide important insight into the genetic determinants contributing to human intelligence.
Similar content being viewed by others


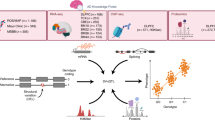
Introduction
Human intelligence, a broad term for mental capabilities involving the ability to reason, plan, solve problems and learn from experience, plays a vital role in educational achievement, career success, and health outcomes [1]. This important construct is commonly measured through tests involving simple concepts like shapes and designs [2]. Intelligence quotient (IQ), a commonly recognized measure of intelligence, is a total score derived from several tests using words, numbers, and specific cultural knowledge [2]. The distribution of IQ is generally considered to be a normal curve centered around an average score of 100, with ~3% of Americans scoring above 130 and nearly the same percent scoring below 70 [2]. Previous studies based on genome-wide complex trait analysis (GCTA) showed that 60% of the phenotypic variation in human intelligence could be accounted for by genetic factors [3].
Although researchers have focused on the identification of genetic determinants related to human intelligence for over a century, the fundamental neural underpinnings of intelligence remain unclear [4]. Genome-wide association studies (GWASs) using meta-analyses can be used to reveal an abundance of trait-associated single nucleotide polymorphisms (SNPs) for many complex phenotypes [5]. One of the largest current GWAS analyses for human intelligence (n = 78,308) identified 336 associated SNPs at the genome-wide significance level located in 18 genomic loci [5]. However, the vast majority of GWAS-reported variants are not biologically causal but are instead correlated with the actual causal variants through linkage disequilibrium (LD) [6].
GWASs provide a crucial and necessary step to interpret the biological mechanisms underlying complex diseases, and to identify novel therapeutic gene targets by prioritizing variants located within those regions. However, due to the complex LD structure, the SNP with the smallest p-value in a given area is not necessarily the most likely to be causal, and it is insufficient to assume that the gene located closest to the top SNP in a given region is the most probable causal gene. Previous studies have shown that the genomic distance of a SNP to a gene does not imply a causal relationship [7]. Due to the presence of LD, it is essential to prioritize the variants within GWAS-associated regions, enabling the conversion of statistical associations into target genes.
Fine-mapping is a statistical analysis approach designed to assign probabilities of causality to candidate variants located in the GWAS-associated regions [8]. This technique may be used to filter the original GWAS findings in order to identify the SNPs that are most likely to be causal for the association signal at a given locus. In recent years, fine-mapping tools have been developed that make use of a variety of algorithms for causal variant identification, including Markov Chain Monte Carlo, exhaustive search, and stochastic search [9]. However, these approaches mainly require individual-level genotype data, which is often difficult to acquire through publicly available sources. Additionally, they generally assume there is only a single causal variant at a given locus, which may not be biologically realistic [10].
To address these issues, several methods have been developed to incorporate GWAS summary statistics into fine-mapping analysis. In particular, the PAINTOR method allows for the integration of the summary statistics, information about the LD structure, and functional annotations to improve the accuracy of causal variant detection [6]. Additional bioinformatics tools based on the GWAS summary statistics include CAVIARBF[10], CAVIAR [11], and FINEMAP [12]. However, these other approaches cannot consider functional annotations or jointly fine-map multiple loci of interest simultaneously.
In this study, we applied the PAINTOR fine-mapping technique to further refine the results from an existing GWAS meta-analysis of human intelligence. We aim for these results to inform future functional validation and molecular biology studies to determine the variants with a true causal effect for the phenotype.
Materials and methods
GWAS dataset
The GWAS summary statistics were acquired from a meta-analysis of 78,308 individuals. The study included whole-genome and whole-exome sequencing of eight separate cohorts including UK Biobank web-based measure (UKB-wb; n = 17,862), UK Biobank touchscreen measure (UKB-ts; n = 36,257, non-overlapping with UKB-wb), CHIC consortium (n = 12,441) and five additional smaller cohorts (n = 11,748) [5]. The dataset consists of Z scores and P values showing the association between human intelligence and over 10 million SNPs.
Defining fine-mapping loci
We first selected the set of SNPs from the original GWAS data that were detected at the genome-wide significance level (p < 5 × 10–8), and then sorted this subset by the chromosomal location and p-value. For each selected SNP we used ANNOVAR [13] to perform a gene-based annotation based on the RefGene database from the UCSC Genome Browser. We defined the top SNP in each associated region as the center of the fine-mapping locus, which included a 100-kilobase window of SNPs (50 kb on each side) around the most significant GWAS hit. Previous studies have shown that LD begins to decay between SNPs that are separated by more than 25 kb, so the choice of a 100 kb width is conservative [14]. For each fine-mapping locus we used PLINK [15] to compute the LD matrix of the pairwise correlations between SNPs based on the 1000 Genomes Phase 3 European (CEU) reference panel.
Functional information
Incorporation of the functional annotations of each SNP can improve the accuracy of causal variant selection [16]. The functional annotations were made available by Gusev et al. [17] in a heritability study and are publicly available through the Broad Institute (Cambridge, MA, USA; https://data.broadinstitute.org/alkes group/). We created a binary (0/1) annotation matrix to assign SNPs to their corresponding functional annotations. The rows of the matrix show the rsID of each SNP, and the columns correspond to the functions of the given SNP, including coding, untranslated region (3′ and 5′ UTR), promoter, DNase hypersensitivity site (DHS) in any of 217 cell types, intron and intergenic [17].
The coding annotation corresponds to the exonic coding regions of the gene that are responsible for protein coding. Untranslated regions (3′ and 5′ UTR) represent the areas of mRNA directly upstream or downstream from the translation initiation codon. Promoters indicate regions of DNA that initiate the transcription of a particular gene. DHS are regions of chromatin that are sensitive to cleavage by the DNase I enzyme and have important implications for gene expression. Finally, introns are the non-protein coding regions that are removed during RNA splicing, while intergenic denotes regions that are located between gene bodies [18].
Statistical analysis
Prior probability
In this study, we used PAINTOR, an expectation maximization algorithm, to compute the posterior probabilities of causality for each SNP at a given fine-mapping locus. This approach incorporates several types of information including the association Z-scores from the summary statistics, LD matrix of pairwise correlation coefficients, and functional annotations [6]. First, we assume Cj is the indicator vector, giving the causal status of all the SNPs in each locus where Cij = 1 if the ith SNP is causal, Cij = 0 otherwise. λj is the vector of non-centrality parameters, and Aijk is the binary indicator of annotations for the ith SNP at locus j in the kth annotation. According to the method, Z-scores for the SNPs at a particular fine-mapping locus follow a multivariate normal distribution. Because of this, we have Eq. (1), where the symbol ο denotes the element-wise multiplication of the two vectors and Σj is the LD matrix including the pairwise Pearson Correlation coefficients between SNPs at jth loci. The prior probability in the algorithm is the effect of each functional element on the likelihood of causality for the SNPs at each locus, produced by PAINTOR through a logistic probability model. For the prior probability, we assume γk as the effect of the kth functional annotation on the probability of causality. We have Eq. (2) for computing the prior probability of the Bayes formulation.
Posterior probabilities
Next, PAINTOR computes posterior probabilities of each causal configuration of SNPs Cj in the total set of all possible causal configurations Qj (\(\left| {Q_j} \right| = \mathop {\sum}\nolimits_{i = 0}^S {\left( {\begin{array}{*{20}{c}} {N_j} \\ i \end{array}} \right)}\), where S is the number of potential causal variants to be considered at a locus and Nj is the total number of SNPs at the jth locus), through the application of Bayes’ rule at each locus independently. With the two equations above, we have Equation (3) to produce posterior probabilities.
To obtain the posterior probability for each SNP at per locus, PAINTOR calculates the possibilities of each causal configuration, shown in Eq. (4).
Likelihood ratio test (LRT) for functional element
To determine whether each functional element influences the posterior probability computation, PAINTOR calculates the log2 relative probability of causality between models incorporating different functional information and performs the LRT to test the statistical significance of the estimated prior probability. According to the PAINTOR method, γ0 is considered as the baseline estimate in a logistic model without any functional annotation. The prior causal probability for any SNP belonging to the kth annotation was estimated through γk. PAINTOR computes the log2 of the ratio of prior causal probability between the kth annotation and baseline to compare the effects between different annotations on causal probability for SNPs. For the LRT, we used Eq. (5) to calculate the likelihoods of each model and compare their log-likelihoods to the base model to determine their significance through Eq. (6). The test statistics asymptotically follow a chi-square distribution with one-degree freedom. When the log-LRT is significant (p < 0.05), the causal variants are significantly enriched in the given functional element indicating that any SNPs residing in this annotation should be assigned the appropriate prior probability that is different from the baseline effect [6].
Results
Fine-mapping loci for human intelligence
The original GWAS results indicate that 336 SNPs are detected at the genome-wide significance level, located at 10 different chromosomes (1, 2, 3, 5, 6, 7, 13, 16, 17, 22). We identified 14 fine-mapping loci based on the genomic locations of the GWAS-associated SNPs. These loci have a mean of 659 SNPs and a standard deviation of 220.0912 (Table 1a, b).
Incorporation of functional annotation data
To improve the accuracy of the fine-mapping analysis, we incorporated the functional annotations of selected SNPs. After matching SNPs with their corresponding functional annotations, we found that the majority of the SNPs are located in the intergenic and intronic regions. In order to calculate the prior probability of causality for each functional element, we built a model containing all annotations of interest, including Coding, UTR, Promoter, DHS, Intron, and Intergenic. There are minimal differences between the prior probabilities estimated for each functional annotation, suggesting that in this fine-mapping analysis, all SNPs are weighted relatively evenly regardless of functional class (Table 2). We also calculated the log2 relative probability of causality for each functional element, which represents the fold enrichment or depletion of causal SNPs within each annotation. However, after performing the LRT for each functional annotation, the results show that the functional information does not significantly influence the analysis (p < 0.05). The causal SNPs are not significantly enriched in any particular functional elements for human intelligence across the genome and including this information does not greatly improve the accuracy of the posterior probability calculation. Thus, the baseline model will be applied to evaluate the posterior probability.
Identifying variant with highest posterior probability
We summarize the posterior probabilities of all SNPs in 14 fine-mapping loci (Supplementary Table 1) and present top five SNPs that are most likely to have a causal effect on human intelligence (Table 3). It is noteworthy that the SNPs with the highest posterior probability of causality are not necessarily the most statically significant SNPs in the original GWAS analysis. There is only one SNP, rs6002620 (NDUFA6-AS1), with posterior probability higher than 0.90. This SNP is the most likely variant to have a causal effect on human intelligence.
Discussion
In this fine-mapping analysis, we incorporated the summary statistics from a publicly available GWAS, information about the pairwise correlations between SNPs, and functional annotations to determine the genomic loci with the highest probability of playing a causal role in human intelligence. This secondary downstream analysis is meant to expand upon the GWAS findings in order to narrow the original results and discern the SNPs that may have the most biological relevance. The exploration of causal variants is a crucial step in understanding the mechanisms underlying complex phenotypes and finding effective ways to prevent and treat human disease. Although there are various proposed methods for performing fine-mapping analysis using GWAS summary statistics, we chose to apply the PAINTOR algorithm since previous simulations indicated that compared with other approaches, PAINTOR showed higher accuracy in selecting causal SNPs [6].
Based on the original summary statistics of human intelligence, we identified 14 fine-mapping loci containing 9226 total SNPs, of which 1323 SNPs reached genome-wide significance. This analysis aims to distinguish the small subset of SNPs that are most likely to be causal. After applying this statistical approach, we selected the top five candidate SNPs with the highest probability of causality for human intelligence. Interestingly, four of the top five SNPs selected are not the most statistically significant SNP located at their respective fine-mapping locus.
It is also important to note that some SNPs with highly significant p-values in the original GWAS analysis do not have high posterior probabilities of causality. While many of the original GWAS hits simply tag the true causal variant, there are also several other possible explanations. For instance, we do not consider topologically associated domains (TADs), which are believed to have an important impact on the functional genome [19]. TADs refer to regions of the genome located far away from each other that may interact [19]. Therefore, it is plausible that the causal variant responsible for a given association signal may reside in a different genomic region that interacts with the GWAS-detected locus. Furthermore, in certain instances the PAINTOR algorithm may be unable to differentiate between SNPs when a given region contains many variants with high levels of statistical significance and a very complex correlation structure.
The two most noteworthy potential causal variants detected for human intelligence are rs6002620 and rs41352752. The SNP rs6002620 has a posterior probability of 0.98 and is located in an intronic region of the gene NDUFA6. The NDUFA6 gene codes for the NADH dehydrogenase 1 alpha subcomplex subunit 6, an accessory subunit of ubiquinone oxidoreductase (Complex I), which is the most abundant enzyme of the mitochondrial membrane respiratory chain [20]. Previous studies performing gene-based analysis of verbal-numerical reasoning have indicated that NDUFA6 is involved in mitochondrial function [21]. Hence, we assume that NDUFA6 may affect the function of neural cells through mitochondrial function, influencing human intelligence. Further biological experiments are needed to prove this assumption.
On the other hand, the SNP rs41352752 has a posterior probability of 0.82 and is located in the intronic region of the gene MEF2C. The gene MEF2C is considered a novel gene associated with human intelligence [21]. Previous studies suggest that MEF2C in cortical pyramidal neurons may regulate synapse densities in early development by acting in the postsynaptic neuron as a cell-autonomous, transcriptional repressor on crucial target genes. Furthermore, knockout of MEF2C is associated with the alteration of gene expression for numerous autism and synapse linked genes, and in turn may lead to diseases related to mental deficiency [22]. Due to the strong association between MEF2C and human psychological deficiencies, we believe that this SNP may be highly important for human intelligence.
Additionally, we highlight the SNP rs6568547, which has a posterior probability of 0.62 and resides in the intergenic region between AFG1L and FOXO3. AFG1L encodes a mitochondrial integral membrane protein that plays a role in mitochondrial protein homeostasis [23]. The evidence suggests that this gene is associated with bipolar disorder, a mental health condition that causes extreme shifts in mood, energy, and behavior [21]. FOXO3 is involved in the preservation of neural stem cells and the development of human intelligence [24]. Although the SNP rs6568547 is located in an intergenic region, the two closest genes are related to human intelligence and mental dysfunction.
Finally, the SNP rs138592330 has a posterior probability of 0.60 and resides in the intergenic region between eIF3C and NPIPB9. The gene eIF3C is one of the three evolutionarily conserved subunits of eIF3 (eIF3a, eIF3b, and eIF3c), which binds to a highly specific program of mRNAs involved in cell growth control processes such as cell cycling, differentiation, and apoptosis [25]. Specifically, this gene may affect the development of the brain cells related to human intelligence. We note that this SNP was not detected by the original GWAS analysis (p = 1.2 × 10–6). This suggests that GWASs may easily miss true causal variants, indicating a need for novel approaches to improve the accuracy of the selection of variants that exert a true biological effect.
A major advantage of PAINTOR is that it can up-weight the SNPs that reside in the functional elements where causal SNPs are enriched. Previous studies have shown that the most significant gain from considering functional information occurs at those loci where the association signal is weaker, and the most critical SNPs have smaller effect sizes [16]. In this analysis, we found that the causal SNPs were not enriched in any particular functional elements, and that the posterior probabilities of the SNPs mainly depend on the Z-scores and the regional correlation structure. In this particular case, the functional elements may not have a substantial effect on the analysis; however, we still believe the accuracy of causal variant detection may be improved by including this information.
There are also several important limitations of this statistical fine-mapping approach. For instance, it is not possible to determine the proportion of variability in human intelligence that is explained by these five candidate causal SNPs without access to the individual-level data. Additionally, this statistical exercise cannot be used for the definitive identification of causal variants without further functional validation. However, we believe this analysis is a crucial step for further understanding the genetic determinants contributing to human intelligence. These results should serve as a reference for molecular biologists in prioritizing the genetic loci for performing function mechanistic experiments related to human intelligence.
In summary, we used a Bayesian approach to determine the SNPs with the highest probability to be causal variants for human intelligence, based on the summary statistics from existing GWAS data. Through the identification of these causal variants, we may be able to gain novel insight into the mechanisms involved in the development of elevated levels of intelligence and pathogenic cognitive conditions.
References
Neisser U, Boodoo G, Bouchard Jr. TJ, Boykin AW, Brody N, Ceci SJ, et al. Intelligence: knowns and unknowns. Am Psychol. 1996;51:77–101.
Gottfredson LS. Mainstream science on intelligence: an editorial with 52 signatories, history, and bibliography. Intelligence. 1997;24:13–23.
Trzaskowski M, Yang J, Visscher PM, Plomin R. DNA evidence for strong genetic stability and increasing heritability of intelligence from age 7 to 12. Mol Psychiatry. 2014;19:380–4.
Coleman JR, Bryois J, Gaspar HA, Jansen PR, Savage JE, Skene N, et al. Biological annotation of genetic loci associated with intelligence in a meta-analysis of 87,740 individuals. Mol Psychiatry. 2018; https://doi.org/10.1038/s41380-018-0040-6.
Sniekers S, Stringer S, Watanabe K, Jansen PR, Coleman JRI, Krapohl E, et al. Genome-wide association meta-analysis of 78,308 individuals identifies new loci and genes influencing human intelligence. Nat Genet. 2017;49:1107–12.
Kichaev G, Yang WY, Lindstrom S, Hormozdiari F, Eskin E, Price AL, et al. Integrating functional data to prioritize causal variants in statistical fine-mapping studies. PLoS Genet. 2014;10:e1004722.
Petersen A, Alvarez C, DeClaire S, Tintle NL. Assessing methods for assigning SNPs to genes in gene-based tests of association using common variants. PLoS ONE. 2013;8:e62161.
Spain SL, Barrett JC. Strategies for fine-mapping complex traits. Hum Mol Genet. 2015;24:R111–9.
Greenbaum J, Deng HW. A statistical approach to fine mapping for the identification of potential causal variants related to bone mineral density. J Bone Min Res. 2017;32:1651–8.
Chen W, Larrabee BR, Ovsyannikova IG, Kennedy RB, Haralambieva IH, Poland GA, et al. Fine mapping causal variants with an approximate Bayesian method using marginal test statistics. Genetics. 2015;200:719–36.
Hormozdiari F, Kichaev G, Yang WY, Pasaniuc B, Eskin E. Identification of causal genes for complex traits. Bioinformatics. 2015;31:i206–13.
Benner C, Spencer CC, Havulinna AS, Salomaa V, Ripatti S, Pirinen M. FINEMAP: efficient variable selection using summary data from genome-wide association studies. Bioinformatics. 2016;32:1493–501.
Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164.
Conrad DF, Jakobsson M, Coop G, Wen X, Wall JD, Rosenberg NA, et al. A worldwide survey of haplotype variation and linkage disequilibrium in the human genome. Nat Genet. 2006;38:1251–60.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75.
Chen W, McDonnell SK, Thibodeau SN, Tillmans LS, Schaid DJ. Incorporating functional annotations for fine-mapping causal variants in a Bayesian framework using summary statistics. Genetics. 2016;204:933–58.
Gusev A, Lee SH, Trynka G, Finucane H, Vihjalmsson BJ, Xu H, et al. Partitioning heritability of regulatory and cell-type-specific variants across 11 common diseases. Am J Hum Genet. 2014;95:535–52.
Costa FF. Non-coding RNAs, epigenetics and complexity. Gene. 2008;410:9–17.
Sexton T, Cavalli G. The role of chromosome domains in shaping the functional genome. Cell. 2015;160:1049–59.
Derambure C, Dzangue-Tchoupou G, Berard C, Vergne N, Hiron M, D’Agostino MA, et al. Pre-silencing of genes involved in the electron transport chain (ETC) pathway is associated with responsiveness to abatacept in rheumatoid arthritis. Arthritis Res Ther. 2017;19:109.
Davies G, Marioni RE, Liewald DC, Hill WD, Hagenaars SP, Harris SE, et al. Genome-wide association study of cognitive functions and educational attainment in UK Biobank (N = 112 151). Mol Psychiatry. 2016;21:758–67.
Harrington AJ, Raissi A, Rajkovich K, Berto S, Kumar J, Molinaro G, et al. MEF2C regulates cortical inhibitory and excitatory synapses and behaviors relevant to neurodevelopmental disorders. eLife. 2016;5:e20059.
Cesnekova J, Rodinova M, Hansikova H, Houstek J, Zeman J, Stiburek L. The mammalian homologue of yeast Afg1 ATPase (lactation elevated 1) mediates degradation of nuclear-encoded complex IV subunits. Biochem J. 2016;473:797–804.
Webb AE, Pollina EA, Vierbuchen T, Urban N, Ucar D, Leeman DS, et al. FOXO3 shares common targets with ASCL1 genome-wide and inhibits ASCL1-dependent neurogenesis. Cell Rep. 2013;4:477–91.
Lee AS, Kranzusch PJ, Cate JH. eIF3 targets cell-proliferation messenger RNAs for translational activation or repression. Nature. 2015;522:111–4.
Acknowledgements
This work was not funded but benefited by the support from the National Institutes of Health [P50AR055081, R01AG026564, R01AR050496, R01AR057049], and Edward G. Schlieder Endowment fund from Tulane University.
Author contributions
Y.G. performed data analysis and produced the manuscript. J.G. provided advice during data analysis and manuscript revisions. H.W.D. initiated and developed the project and oversaw the application of statistical methods.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Gong, Y., Greenbaum, J. & Deng, HW. A statistical approach to fine-mapping for the identification of potential causal variants related to human intelligence. J Hum Genet 64, 781–787 (2019). https://doi.org/10.1038/s10038-019-0623-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s10038-019-0623-3
This article is cited by
-
Identification and validation of a regulatory mutation upstream of the BMP2 gene associated with carcass length in pigs
Genetics Selection Evolution (2021)
-
Genome-wide association study identifies new loci associated with noise-induced tinnitus in Chinese populations
BMC Genomic Data (2021)
