
Abstract
The genetic causes of Leigh syndrome are heterogeneous, with a poor genotype–phenotype correlation. To date, more than 50 nuclear genes cause nuclear gene-encoded Leigh syndrome. NDUFS6 encodes a 13 kiloDaltons subunit, which is part of the peripheral arm of complex I and is localized in the iron-sulfur fraction. Only a few patients were reported with proven NDUFS6 pathogenic variants and all presented with severe neonatal lactic acidemia and complex I deficiency, leading to death in the first days of life. Here, we present a patient harboring two NDUFS6 variants with a phenotype compatible with Leigh syndrome. Although most of previous reports suggested that NDUFS6 pathogenic variants invariably lead to early neonatal death, this report shows that the clinical spectrum could be larger. We found a severe decrease of NDUFS6 protein level in patient’s fibroblasts associated with a complex I assembly defect in patient’s muscle and fibroblasts. These data confirm the importance of NDUFS6 and the Zn-finger domain for a correct assembly of complex I.
Similar content being viewed by others

Introduction
Mitochondrial complex I in mammals is composed of 45 subunits encoded by both the mitochondrial and nuclear genomes [1]. Mature mitochondrial complex I is associated with respiratory complexes III and IV to form respiratory chain supercomplexes [2,3,4]. The NDUFS6 gene encodes a 13 KDa subunit, which is part of the peripheral arm and is localized in the iron-sulfur fraction of the complex I.
Complex I deficiency is associated with a wide range of clinical presentations, including marked and often fatal lactic acidosis, cardiomyopathy, leukoencephalopathy, pure myopathy, and hepatopathy with tubulopathy [5]. Complex I deficiency is the most frequently observed abnormality and accounts for ~30% of the cases of Leigh syndrome [6].
The symptoms of Leigh syndrome are highly variable, but usually include psychomotor arrest or regression, hypotonia, dystonia, seizures, ocular movements, and respiratory failure. Onset is typically between 3 and 12 months; about 50% of affected individuals die by 3 years of age. Biochemically, elevated lactate levels in the blood and cerebral spinal fluid are frequently encountered. The genetic causes of Leigh syndrome are heterogeneous, with a poor genotype–phenotype correlation. To date, >50 nuclear genes cause nuclear gene-encoded Leigh syndrome [7]. Diagnostic rates have improved considerably following the adoption of next-generation sequencing (NGS) technologies, but are still far from perfect. Even using whole-exome sequencing, around 50% of cases remain without molecular diagnosis [8]. To improve this rate, novel approaches using phenomic annotation have been described, highlighting the challenges associated with integrating phenomic technologies into clinical practice [9]. Moreover, Ogawa et al. [8] demontrasted the importance of combining multiple methods of diagnosing Leigh/Leigh-like patients. Combining genetic analysis and enzyme assay, mitochondrial respiratory chain defect were confirmed in 82% of cases.
To date, only a few patients were reported with NDUFS6 pathogenic variants. Most of them presented with severe neonatal lactic acidemia and complex I deficiency leading to death in the first days of life [10,11,12,13] (Table 1). Recently, Ogawa et al. [8] reported a patient with Leigh syndrome who harbored two novel NDUFS6 variants, c.309 + 5 G > A and c.343 T > C (p.Cys115Arg). However, they did not report any detailed clinical information and the pathogenicity of both variants was not functionally proven. Here, we provide a clinical and molecular description of another patient, bearing the same NDUFS6 variants and presenting with a phenotype compatible with Leigh syndrome. We show that the two variants lead to a disassembly of complex I in patient’s muscle and fibroblasts and explain why those patients exhibit a relatively milder phenotype.
Materials and methods
Case report
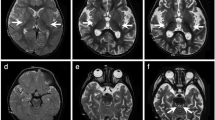
The proband was born at term following uneventful pregnancy of healthy unrelated parents of French origin. He had one healthy older sister. After birth, he presented a congenital torticollis, edema of the eyelid during 15 days, and feeding difficulties with significant gastroesophageal reflux. Head control was acquired at 4 months. A bilateral divergent strabismus and nystagmus were then observed, without anomaly associated at ophthalmological examination. Brain MRI (magnetic resonance imaging) performed at 5 months of age was normal (Fig. 1a, b). At 6 months, neurological examination showed a central hypotonia with axial hypotonia and peripheral hypertonia and a poor maintenance of eye contact. At 7 months, he exhibited a sudden neurological deterioration, during a viral infection with, progressively over a few days, many episodes of unconsciousness with severe hypotonia and respiratory disorders, evolving to a coma with hypoventilation, requiring endotracheal intubation, and persistent high blood pressure. After correction of the respiratory acidosis, the biological analyses showed a lactic acidosis with hyperlactatemia (4.73 mmol/L, n: 0.6–2.2 mmol/L), elevated lactate/pyruvate ratio (27, n < 10) and hyperlactatorachia (2.77 mmol/L, n: 1.2–2 mmol/L). Brain MRI showed symmetric lesions of corticospinal tracts into the medulla (bulbo–protuberantial junction) with hyperintensity T2-weighted images (Fig. 1c, d) and hypointensity T1 (Fig. 1e) and mild hyperintensity T2 of globi pallidi (Fig. 1f).

Brain magnetic resonance imaging. Normal brain MRI performed at 5 months of age (a, b). At 7 months of age, symmetric lesions of corticospinal tracts into medulla (bulbo–protuberantial junction) with hyperintensity T2-weighted images (c, d), hypointensity T1 (e) and mild hyperintensity T2 of globi pallidi (f). Lesions are shown with arrows
Patient derived fibroblasts were examined for the activity of the respiratory chain enzymes by spectrophotometric analysis, which did not show any defect (Fig. 3a), while spectrophotometric analysis of patient’s muscle revealed a defect in complexes II and IV activities compared to controls (Fig. 3b).
His evolution was afterwards slowly favorable. He was able to play with toys and to hold his head but axial hypotonia and peripheral hypertonia persisted with occurrence of a bilateral ophtalmoplegia. At 11 months, a new sudden neurological deterioration occurred leading to his death.
Custom targeted panel analysis
Informed consent for diagnostic and research studies was obtained from proband’s parents in accordance with the Declaration of Helsinki protocols. We designed a custom panel of genomic regions corresponding to 281 genes, known in 2016 to be involved in mitochondrial disorders. Sequencing and filtering were performed as described before [14]. In short, we designed RNA probes to capture the transcribed sequences of genes with Agilent SureSelect kit (Agilent, Santa Clara, California, USA). Ion PI chips V3 were sequenced on the Ion Proton, using Ion PI Hi-Q sequencing kit (Thermo Fisher, Foster City, CA, USA). The sequences were mapped to the hg19 reference genome and variant calling was then performed using variant caller version 5.0.4.0. Common variants were filtered out based on frequency of < 1% in the 1000 genomes project or in the 5000 exomes european-american (NHLBI ESP). We focused on predicted missense, frame-shift, stop-gain or stop-loss, and splice-site variants. Variants identified by NGS were validated by Sanger sequencing.
NDUFS6 Sanger sequencing
Amplicons were amplified by using standard PCR conditions with 5′-GGTGGAGACTCGGGTGATAG-3′ forward primer and 5′-CGGGAGCAGGTAGAGAAGAG-3′ reverse primer for exon 3, and 5′-GAGATCGACAGTCTGCATTCT-3′ forward primer and 5′-CACAGAACCAGCGAGCTTC-3′ reverse primer for exon 4. PCR products were purified with Illustra ExoProStar enzyme (GE Healthcare, Little Chalfont, England), processed with a BigDye® Terminator Cycle Sequencing kit (Thermo Fisher, Foster City, CA, USA) and analyzed on an ABI3130XL automated sequencer (Thermo Fisher).
Reverse transcription PCR (RT-PCR) analysis
Total RNAs were extracted by using the Trizol reagent (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. Complementary DNA (cDNA) synthesis was performed using the Kit Transcriptor first strand cDNA Synthesis (Roche Diagnostics, Mannheim, Germany) and was followed by PCR amplification using 5′-AAAGGCCAGCGGCGCAAA-3′ forward primer and 5′-ACACCCTTTATTCAGCACCAG -3′ reverse primer.
Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), blue native polyacrylamide gel electrophoresis (BN-PAGE)
SDS-PAGE using a 12% SDS gel and BN-PAGE using a 4–13% acrylamide gradient gel were performed as previously described [15].
The following primary antibodies were used: NDUFS6 (Abcam, Cambridge, UK; ab195807), NDUFA13/GRIM19 and NDUFB6 for complex I (Abcam; ab110240 and ab110244, respectively), SDHA for complex II (Abcam; ab14715), UQCRC2 for complex III (Abcam; ab14745), MTCO1 for complex IV (Abcam; ab14705), ATPB for complex V (Abcam; ab14730), a cocktail of anti-human total OXPHOS complex antibodies (Mitosciences, Eugene, Oregon, USA), and GRIM19 subunit of complex I, SDHA subunit of complex II, UQCRC2 subunit of complex III, MTCO1 subunit of complex IV and ATP5A subunit of complex V (Mitosciences) for BN-PAGE analysis.
OXPHOS spectrophotometric measurements
Enzymatic spectrophotometric measurements of the OXPHOS (oxidative phosphorylation) respiratory chain complexes and citrate synthase were performed at 37 °C on patient’s muscle according to standard procedures using a spectrophotometer Cary50 (Varian) [16]. Proteins were measured according to Bradford microassay [17].
Results
Identification of two NDUFS6 variants
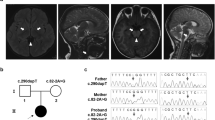
A pathogenic variant of mitochondrial DNA was first ruled out by NGS sequencing from muscle sample (Table sup data). Then, sequencing of a panel of 281 nuclear genes involved in mitochondrial disorders led to the identification of two variants of the NDUFS6 gene: c.343 T > C (p.Cys115Arg) and c.309 + 5 G > A. Both variants were previously identified in a patient presenting with Leigh syndrome [8]. However, their pathogenic effect was not functionally proven. The c.343 T > C was inherited from the father while the c.309 + 5 G > A was inherited from the mother confirming that our proband is compound heterozygous for both variants (Fig. 2a, b). The c.343 T > C substitution changes a highly conserved cysteine (Cys) into an arginine (Arg) at amino acid position 115 (Fig. 2c). A pathogenic substitution was previously described at this position changing the cysteine into a tyrosine instead [10]. The c.343 T > C variant was not found in the Genome Aggregation Database (GnomAD) (http://gnomad.broadinstitute.org/) and was predicted to be “disease causing” based upon in silico analysis with SIFT (http://sift.jcvi.org/), PolyPhen2 (http://genetics.bwh.harvard.edu/pph2/), and Mutation Taster (http://www.mutationtaster.org/).

Identification of NDUFS6 variants. a Pedigree of the proband (II-2). b Sanger sequencing showing c.309 + 5 G > A (upper panel) and c.343 T > C (lower panel) variants in the proband. c Interspecies amino acid comparison showing that the cysteine (c) at position 115 is highly conserved (red box). d Cryo-EM structure of human respiratory complex I matrix arm (PDB code: 5XTB), ISC: iron–sulfur clusters. The NDUFS6 protein (blue) and the residue Cys 115 (red) are indicated. e RT-PCR and sequencing analysis for the c.309 + 5 G > A variant. The genomic DNA segment tested is shown schematically above the panel. E1 to E4 represent exons of the NDUFS6 gene. On the left, agarose gel loaded with RT-PCR products from control (C) and proband (P) using forward primer in exon 1 and reverse primer in exon 4. The sequence chromatograms of the two bands from the proband’s RT-PCR products are shown (*wild type allele, **mutated allele). Splicing patterns are shown schematically alongside the right-hand margin of the sequences. M, molecular weight marker; P, patient; C, control subject; (-): negative PCR control. f RT-PCR and sequencing analysis using a forward primer spanning the exon 3/exon 4 boundary, and a reverse primer in exon 4
Cryo-EM structure of human respiratory complex I matrix arm (PDB code: 5XTB) was used to analyze the functional impact of the p.Cys115Arg variant. The cysteine residue at position 115 is part of the Zn-binding site needed for assembly and stability of complex I. The p.Cys115Arg substitution is predicted to alter the interactions necessary for proper complex I assembly and function (Fig. 2d).
The c.309 + 5 G > A variant changes a highly conserved nucleotide closed to the canonical splice donor site in intron 3. This base substitution was reported only once in GnomAD. It was predicted to drastically decrease the intrinsic strength of the exon 3 donor splice-site by using the splice-site prediction algorithm Human Splicing Finder (http://www.umd.be/HSF/). The Sanger sequencing of the entire NDUFS6 cDNA with a forward primer in exon 1 and a reverse primer in the last exon (exon 4) showed an aberrantly spliced mRNA, missing exon 3, in the patient compared with the control (Fig. 2e). We also checked if the c.309 + 5 G > A variant could leave some amount of normally spliced product. We sequenced the cDNA of our patient using a forward primer 5′-GTATATAAACTTGGACAAAGAAACA-3′, spanning the exon 3/exon 4 boundary, and a reverse primer in exon 4, 5′-CCCTTTATTCAGCACCAGGA-3′. We observed the variant c.343 C but also, in a smaller proportion, the wild-type allele c.343 T (Fig. 2f). This result suggests that a small proportion of mRNA is normally spliced and allows a certain proportion of normal NDUFS6 protein to be translated and in fine a normally functioning complex I.
NDUFS6 variants lead to decrease of NDUFS6 level and complex I assembly defect
In patient’s fibroblasts, we observed a clear decrease of NDUFS6 level compared with the control by western-blot analysis confirming the impact of both variants on the NDUFS6 expression (Fig. 3c).

OXPHOS, BN-PAGE, and western-blot analysis in muscle and fibroblasts of the patient. a, b Mitochondrial respiratory enzyme activities in fibroblasts (a) and in muscle (b) from patient and controls. The decreased values are in bold. c Western blot performed with a NDUFS6 antibody in fibroblasts from patient (P) and control individual (C). GAPDH and Hsp60 were used as loading controls. d, e BN-PAGE performed in fibroblasts (d) and in muscle (e) from patient (P) and control individual (C)
BN-PAGE analysis, with the GRIM19/NDUFA13 antibody, showed an assembly defect of complex I in fibroblasts (Fig. 3d) and in muscle (Fig. 3e). However, in fibroblasts, we observed a small amount of fully assembled complex I. We also performed BN-PAGE analysis, with another antibody against NDUFB6 subunit, which confirmed the loss of fully assembled complex I in the patient’s muscle compared with control (Fig. 3e). No assembly intermediates could be observed.
Discussion
Spiegel and Kirby have first described children with pathogenic variants in the NDUFS6 gene [10, 11]. All of them died in the first month of life. Later, other patients with lethal infantile mitochondrial disease were reported [13, 18] (Table 1). Those patients harbored frame-shift NDUFS6 variants. Only one patient, with an apparently progressive course of the disease and muscular hypotonia, was reported [12]. This patient harbored a homozygous nonsense mutation. More recently, Ogawa et al. [8] reported a study of 106 Japanese patients with Leigh syndrome, including one patient who harbored both c.309 + 5G>A and c.343T>C (p.Cys115Arg) NDUFS6 variants. However, we had no detailed clinical information about this patient and the pathogenicity of both variants was not functionally proven. Herein, we gave a full clinical description of one more patient presenting with Leigh syndrome and harboring both c.309 + 5G>A and c.343T>C (p.Cys115Arg) NDUFS6 variants. This presentation is milder than the neonatal lethal forms previously reported, yet it is clearly associated with serious neurological manifestations and a grave prognosis. Moreover, although the neuropathology of a previously reported proband could be suggestive of Leigh syndrome with prominent congestion of the basal ganglia, thalamus, and periventricular tissue of the brainstem, the lesions were also compatible with severe hypoxia and typical lesions of Leigh disease such as vascular proliferation, loosening of the neuropil, and preservation of neurons were not observed [19]. In our proband, the first clinical signs appeared during the second month of life and both clinical evolution and brain MRI were compatible with Leigh syndrome. Interestingly, brain MRI exhibited prominent bulbo–protuberantial junction lesions in correlation with the clinical signs of brainstem dysfunction observed, and basal ganglia involvement was less prominent than classically observed in Leigh syndrome. Our report underlines the neurological phenotype caused by NDUFS6 while confirming Leigh syndrome to be part of its spectrum.
We have strong arguments for the pathogenicity of both identified NDUFS6 variants. The c.309 + 5G>A variant induces an abnormal splicing between exons 2 and 4, leading to exon 3 skipping. Concerning the c.343T>C (p.Cys115Arg) variant, its highly conservation, in silico predictions and its localization are clearly in favor of its pathogenicity. NDUFS6 is part of the zf-CHCC domain (location :82 → 119) and has a C-terminal CX8–9HX14–15CX2C zinc finger motif. This motif has been shown to be essential for zinc binding in NUMM, the Y. lipolytica ortholog of NDUFS6 [20]. The integrity of the Zn-binding site is needed for proper assembly and stability of complex I. The single Zn(II) ion is coordinated by three cysteine residues, including Cys 128 which corresponds to human NDUFS6 Cys 115, and one histidine. The Zn is in close vicinity of two Fe-S clusters. Interestingly, it has been shown that cluster N4 is missing from complex I in mutant C128A [20]. In human, mutations leading to the exchange of one of the three conserved cysteine residues were shown to cause fatal diseases [10, 11]. So we can assume that, as for Cys 128 in the Y. lipolytica ortholog, Cys 115 in the human NDUFS6 protein could be essential for stable insertion of Fe-S cluster N4 and thus for proper complex I assembly and function. Our results support this hypothesis by showing complex I disassembly in the patient’s muscle and fibroblasts.
Surprisingly, differing from the previous reported NDUFS6 defects, in the patient’s fibroblasts, we did not observed any defect, while spectrophotometric analysis of patient’s muscle revealed combined OXPHOS defect that included complex II and IV. Most of previous reported patients had isolated complex I deficiency [10, 11] and one patient had a combined complex I and IV deficiency [8]. Multiple deficiencies of OXPHOS complexes have been described before in patients with Leigh syndrome and mutations in complex I subunits genes. Complex III was mostly involved but decrease of complex II, complex IV and complex I + III and II + III activities were also observed [8, 21,22,23,24,25]. Complex IV deficiency could be explained by the interaction between complex I and complexes III and IV to form supercomplexes. Indeed, the lack of complex I has been shown to lead to a decrease of the I/III2 and I/III/IVn supercomplexes [2, 25,26,27]. However, complex II is the only enzyme of the respiratory chain, which does not associate with the other respiratory complexes and complex II deficiency could not be explained by supercomplexes destabilization. As we observed this combined defect only in the muscle, it could correspond to a secondary mitochondrial dysfunction, not directly linked to the complex I assembly defect, but caused by additional factors including damage induced by the environment or somatic mutations in modifier genes, which can either exacerbate or compensate for the primary mutation. Indeed, there is a fine line between primary and secondary dysfunction, with a significant overlap, and many articles underlined the difficulty to distinguish primary and secondary mitochondrial dysfunction [28]. Furthermore, even similar missense variants in one and the same complex I subunit have been shown to differentially affect complex I assembly and lead to a spectrum of deficiency rather than an identical cellular phenotype [25].
The deleterious effect on complex I is demonstrated by BN-PAGE analysis showing an assembly defect, like previously described by Kirby et al. [11], in two patients harboring a large homozygous deletion and a homozygous splicing variant, leading to the loss of the NDUFS6 protein. However, in our proband, we observed a residual amount of NDUFS6 protein by western-blot and a small proportion of fully assembled complex I by BN-PAGE in fibroblasts. Furthermore, given that both patients with Leigh syndrome harbored the same c.309 + 5 G > A variant, we checked if the milder clinical phenotype observed, could be explained by a residual amount of normally spliced mRNA. We sequenced the normally spliced mRNA using a primer spanning the exon 3/exon 4 boundary and we could observe the mutated c.343 C variant, located in exon 4, but also, in a smaller proportion, the wild type allele c.343T. In accordance with the western-blot and BN-PAGE results, it suggests that a small proportion of mRNA is normally spliced and allows a normally functioning complex I in basal conditions.
Moreover, Ogawa et al. [8] described that inconsistencies between materials have been frequently observed in nuclear DNA mutated cases and relatively high numbers of enzyme assays return negative results in genetically verified cases of Leigh or Leigh-like syndrome [27]. All together, these reports highlight that biochemical defects can be misleading and imply that a normal result in muscle and/or fibroblast cell line does not exclude the possibility of a primary mitochondrial disorder. Our report also highlights the interest of BN-PAGE analysis in diagnosis to help in genotype/phenotype correlation.
In conclusion, this report expands the clinical spectrum associated with NDUFS6 pathogenic variants, highlight the difficulty to establish phenotype–genotype correlation and the importance of functional tests. Finally, these data confirm that the presence of NDUFS6 and the integrity of the Zn-finger domain are essential for correct assembling of complex I.
References
Brandt U. Energy converting NADH:quinone oxidoreductase (complex I). Annu Rev Biochem. 2006;75:69–92.
Guerrero-Castillo S, Baertling F, Kownatzki D, Wessels HJ, Arnold S, Brandt U, et al. The assembly pathway of mitochondrial respiratory chain complex I. Cell Metab. 2017;25:128–39.
Acin-Perez R, Enriquez JA. The function of the respiratory supercomplexes: the plasticity model. Biochim Biophys Acta. 2014;1837:444–50.
Acín-Pérez R, Fernández-Silva P, Peleato ML, Pérez-Martos A, Enriquez JA. Respiratory active mitochondrial supercomplexes. Mol Cell. 2008;32:529–39.
Loeffen JL, Smeitink JA, Trijbels JM, Janssen AJ, Triepels RH, Sengers RC, et al. Isolated complex I deficiency in children: clinical, biochemical and genetic aspects. Hum Mutat. 2000;15:123–34.
Fassone E, Rahman S. Complex I deficiency: clinical features, biochemistry and molecular genetics. J Med Genet. 2012;49:578–90.
Rahman S, Thorburn D. Nuclear gene-encoded leigh syndrome overview. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Stephens K, et al., éditeurs. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993 [cité 24 févr 2019]. Disponible sur: http://www.ncbi.nlm.nih.gov/books/NBK320989/.
Ogawa E, Shimura M, Fushimi T, Tajika M, Ichimoto K, Matsunaga A, et al. Clinical validity of biochemical and molecular analysis in diagnosing Leigh syndrome: a study of 106 Japanese patients. J Inherit Metab Dis. 2017;40:685–93.
Rahman J, Rahman S. The utility of phenomics in diagnosis of inherited metabolic disorders. Clin Med (Lond). 2019;19:30–6.
Spiegel R, Shaag A, Mandel H, Reich D, Penyakov M, Hujeirat Y, et al. Mutated NDUFS6 is the cause of fatal neonatal lactic acidemia in Caucasus Jews. Eur J Hum Genet. 2009;17:1200–3.
Kirby DM, Salemi R, Sugiana C, Ohtake A, Parry L, Bell KM, et al. NDUFS6 mutations are a novel cause of lethal neonatal mitochondrial complex I deficiency. J Clin Invest. 2004;114:837–45.
Haack TB, Madignier F, Herzer M, Lamantea E, Danhauser K, Invernizzi F, et al. Mutation screening of 75 candidate genes in 152 complex I deficiency cases identifies pathogenic variants in 16 genes including NDUFB9. J Med Genet. 2012;49:83–9.
Swalwell H, Kirby DM, Blakely EL, Mitchell A, Salemi R, Sugiana C, et al. Respiratory chain complex I deficiency caused by mitochondrial DNA mutations. Eur J Hum Genet. 2011;19:769–75.
Plutino M, Chaussenot A, Rouzier C, Ait-El-Mkadem S, Fragaki K, Paquis-Flucklinger V, et al. Targeted next generation sequencing with an extended gene panel does not impact variant detection in mitochondrial diseases. BMC Med Genet. 2018;19:57.
Bannwarth S, Ait-El-Mkadem S, Chaussenot A, Genin EC, Lacas-Gervais S, Fragaki K, et al. A mitochondrial origin for frontotemporal dementia and amyotrophic lateral sclerosis through CHCHD10 involvement. Brain août. 2014;137(Pt 8):2329–45.
Rustin P, Chretien D, Bourgeron T, Gérard B, Rötig A, Saudubray JM, et al. Biochemical and molecular investigations in respiratory chain deficiencies. Clin Chim Acta. 1994;228:35–51.
Bradford M. A rapid and sensitive method for the quantitation of micrograms quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248-54.
Pronicka E, Piekutowska-Abramczuk D, Ciara E, Trubicka J, Rokicki D, Karkucińska-Więckowska A, et al. New perspective in diagnostics of mitochondrial disorders: two years’ experience with whole-exome sequencing at a national paediatric centre. J Transl Med. 2016;14:174.
Rahman S, Blok RB, Dahl HH, Danks DM, Kirby DM, Chow CW, et al. Leigh syndrome: clinical features and biochemical and DNA abnormalities. Ann Neurol. 1996;39:343–51.
Kmita K, Wirth C, Warnau J, Guerrero-Castillo S, Hunte C, Hummer G, et al. Accessory NUMM (NDUFS6) subunit harbors a Zn-binding site and is essential for biogenesis of mitochondrial complex I. Proc Natl Acad Sci USA. 2015;112:5685–90.
Hoefs SJG, van Spronsen FJ, Lenssen EWH, Nijtmans LG, Rodenburg RJ, Smeitink JAM, et al. NDUFA10 mutations cause complex I deficiency in a patient with Leigh disease. Eur J Hum Genet. 2011;19:270–4.
Ortigoza-Escobar JD, Oyarzabal A, Montero R, Artuch R, Jou C, Jiménez C, et al. Ndufs4 related Leigh syndrome: A case report and review of the literature. Mitochondrion. 2016;28:73–8.
Minoia F, Bertamino M, Picco P, Severino M, Rossi A, Fiorillo C, et al. Widening the Heterogeneity of Leigh Syndrome: Clinical, Biochemical, and Neuroradiologic Features in a Patient Harboring a NDUFA10 Mutation. JIMD Rep. 2017;37:37–43.
Angebault C, Charif M, Guegen N, Piro-Megy C, Mousson de Camaret B, Procaccio V, et al. Mutation in NDUFA13/GRIM19 leads to early onset hypotonia, dyskinesia and sensorial deficiencies, and mitochondrial complex I instability. Hum Mol Genet. 2015;24:3948–55.
Baertling F, Sánchez-Caballero L, van den Brand M a M, Fung C-W, Chan SH-S, Wong VC-N, et al. NDUFA9 point mutations cause a variable mitochondrial complex I assembly defect. Clin Genet. 2018;93:111–8.
Budde SM, van den Heuvel LP, Janssen AJ, Smeets RJ, Buskens CA, DeMeirleir L, et al. Combined enzymatic complex I and III deficiency associated with mutations in the nuclear encoded NDUFS4 gene. Biochem Biophys Res Commun. 2000;275:63–8.
Sofou K, De Coo IFM, Isohanni P, Ostergaard E, Naess K, De Meirleir L, et al. A multicenter study on Leigh syndrome: disease course and predictors of survival. Orphanet J Rare Dis. 2014;9:52.
Niyazov DM, Kahler SG, Frye RE. Primary mitochondrial disease and secondary mitochondrial dysfunction: importance of distinction for diagnosis and treatment. Mol Syndromol. 2016;7:122–37.
Acknowledgements
We thank Gaëlle Auge, Mathieu Berthet, Christelle Camuso, Bernadette Chafino, Charlotte Cochaud, and Sandra Foustoul for technical help.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Rouzier, C., Chaussenot, A., Fragaki, K. et al. NDUFS6 related Leigh syndrome: a case report and review of the literature. J Hum Genet 64, 637–645 (2019). https://doi.org/10.1038/s10038-019-0594-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s10038-019-0594-4
This article is cited by
-
Application of the BACs-on-Beads assay for the prenatal diagnosis of chromosomal abnormalities in Quanzhou, China
BMC Pregnancy and Childbirth (2021)
