
Abstract
Obesity has result in increased prevalence of type 2 diabetes (T2D) in children. The genetic mechanisms underlying their relationship, however, are not fully understood. Here, we aim to identify novel SNPs associated with T2D and childhood obesity (CO), especially their pleiotropic loci. We integrated the summary statistics for two independent GWASs of T2D (n = 149,821) and childhood body mass index (CBMI) (n = 35,668) using the pleiotropy-informed conditional false discovery rate (cFDR) method. By leveraging the information of different levels of association for CBMI, we observed a strong enrichment of genetic variants associated with T2D. We identified 139 T2D-associated SNPs with 125 novel ones (cFDR < 0.05). Conditioned on T2D, we identified 37 significant SNPs for CBMI (cFDR < 0.05), including 25 novel ones. The conjunctional cFDR (ccFDR) analysis showed ten novel pleiotropic loci for T2D and CBMI (ccFDR < 0.05). Interestingly, the novel SNP rs1996023 is located at protein coding gene GNPDA2 (ccFDR = 1.28E-02), which has been reported to influence the risk of T2D and CO through central nervous system. Our findings may help to explain a greater proportion of the heritability for human traits and advance the understanding of the common pathophysiology between T2D and CO.
Similar content being viewed by others
Introduction
Type 2 diabetes (T2D) is a progressive disorder characterized by aberrant metabolism of fat and protein and chronic hyperglycemia resulting from deficiency of insulin action [1]. During the past decade, the prevalence of T2D has increased dramatically and it has become a serious global public health issue. In 2014, T2D caused more than 4.9 million deaths over the world [2] and 439 million people may suffer T2D by 2030 [3]. It has become one of the major causes of disability and the rising cost of health care.
Clinical and epidemiological studies showed that experiences in early life (e.g., childhood obesity (CO)), may have long-term impact on adult health [4]. Several groups have demonstrated that CO is associated with increased risk of T2D development in later life. For instance, a prospective cohort study including 2,294,139 Israeli adolescents illustrated that participants suffer from overweight and obesity had significantly increased susceptibility to T2D than those with referential body mass index (BMI) [5]. A recent meta-analysis of 26 independent cohorts showed that high childhood body mass index (CBMI) results in an increased incidence of T2D in later life (Odds ratio = 1.70; 95% CI = 1.30–2.22) [6].
Genome-wide association studies (GWASs) have revealed a number of single-nucleotide polymorphisms (SNPs) for T2D and CO, and common genetic variants were reported in both traits [7]. However, these SNPs only account for 5% and 1.5% heritability for T2D and CBMI, respectively [8, 9], and few studies directly tackled the challenge to identify common shared pleiotropic genes of T2D and CO. New information on the nature of the genetic foundations underlying these highly correlated phenotypes should be investigated. Recently, several innovative analytical techniques [10, 11] were developed to identify the missing novel variants/genes, especially the novel pleiotropic loci associated with both T2D and CO. The pleiotropy-informed conditional false discovery rate (cFDR) method [12], which integrated independent GWASs with summary statistics, could increase the power in detection of genetic variants and elucidate mechanism of genetic relationships between related phenotypes. The application of this approach has successfully identified genetic overlapping between several diseases and phenotypes. For example, Desikan et al. have innovatively reported novel variants associated with Alzheimer’s disease and Parkinson’s disease etc. [13].
In the present study, we integrated two independent GWASs with summary statistics of T2D and CBMI using cFDR method to identify novel genetic loci for these phenotypes and test whether T2D shares common susceptibility loci with CO [14, 15]. Our study gains insights into the genetic mechanism of these correlated phenotypes and highlights the potential common pathophysiology between T2D and CO.
Methods
GWAS datasets
We downloaded two independent GWASs with summary statistics for T2D and CBMI from publicly available datasets. The Diabetes Genetics Replication and Meta-analysis (DIAGRAM) consortium dataset (http://diagram-consortium.org/downloads.html), which currently is the largest meta-analysis study for T2D (n = 149,821), involves 34,840 cases and 114,981 controls, overwhelmingly of European descent. The Early Growth Genetics (EGG) Consortium dataset (http://egg-consortium.org/childhood-bmi.html) represents a collaborative effort to combine over 20 European-based GWASs (n = 35,668) to identify additional genetic variation that have an importance on a variety of traits related to early growth. These two datasets include p-values and effects direction at up to 2.5 million directly genotyped or imputed SNPs using CEU samples from the International HapMap Project (Phase 2, release 22). There is no overlap between subjects in these two datasets. The detailed inclusion/exclusion criteria and characteristics of phenotype in each GWAS are described in the original publications [14, 15].
Data processing
The data processing was performed as described in our previous study [16], we annotated 92,752 shared SNPs by integrating two GWAS Meta-analysis summary statistics, then calculated linkage disequilibrium (LD) between each pair of SNPs in a window of 50 SNPs. If pairwise SNPs have R2 > 0.2, then we removed the SNP with the smaller minor allele frequency. Following this initial SNP removing process, we shifted the window in each five SNPs forwardly and repeated this procedure until there was no pairwise SNPs in high LD. The HapMap 3 genotypes were used during the pruning procedure. In total, there were 31,209 variants remaining to be used in the cFDR analysis. Genomic control corrects for population structure by adjusting GWAS statistics at each SNP using a uniform overall inflation factor [17]. This procedure was previously processed by the original publications [14, 15], therefore we did not repeat and reapply this correction in this study.
Statistical analysis
The evaluation of pleiotropic enrichment and genetic correlation
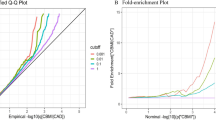
The quantile–quantile plot (Q–Q plot) is a common approach for GWAS to show the observed values (y-axis) comparing to the expected distribution of test statistics (x-axis) across millions of SNPs. The deviation from the x = y line indicates either an incorrect assumed distribution or true associations across the whole genome. To evaluate the pleiotropic enrichment of association, we constructed conditional Q–Q plots as Andreasson et al. [12] mentioned by continuously conditioning the principal trait on SNPs across different level of significance threshold for the conditional trait. Specifically, we plot quantiles of empirical −log10(q) values on x-axis and quantiles of nominal −log10(p) values on y-axis for T2D and CO, respectively. The strength of pleiotropy enrichment was evaluated from the degree of leftward shift from line x = y as the principal trait is successively conditioned on more stringent significance thresholds in the conditional trait.
Meanwhile, we used linkage disequilibrium score regression (LDSC) for estimating genetic correlation from GWAS summary statistics [18, 19]. We followed the instructions (https://github.com/bulik/ldsc/) and performed this analysis in python 3 software.
The calculation of the cFDR
We employed cFDR method [12], an revision of the FDR approach, to determine the novel loci for T2D and CO. The detailed procedures were given by Desikan et al. [12]. Generally, this approach integrated independent GWASs with summary statistics to assess whether a random SNP is related to the principal phenotype given the observed p-values for two traits are smaller than the pre-defined disease-specific significance levels [20]. Specifically, we calculated the cFDR for each SNP where principal trait T2D is conditioned on the association with CO (T2D|CO) and vice versa (CO|T2D). To test the enrichment of the specific loci, we successively confined the strata of SNPs being assessed according to the significance level for the association of each SNP with the conditional phenotype under the following threshold: p-value < 1 for all SNPs, p-value < 0.1, p-value < 0.01, and p-value < 0.001. A significance threshold of 0.05 was applied to distinguish the principal trait associated SNPs. The cFDR Manhattan plot was used to illustrate the chromosomal location of various significant SNPs, which was associated with T2D conditioned the association with CO and vice versa.
The calculation of conjunctional cFDR (ccFDR)
We computed the ccFDR, the maximum cFDR values (i.e., T2D|CO and CO|T2D) of both the principal trait and conditional trait, to assess the pleiotropic loci where a given SNP has a false positive association with the two phenotypes. The threshold for a significant pleiotropic locus was defined as ccFDR of 0.05. To visualize the chromosomal position of identified pleiotropic loci, we constructed ccFDR Manhattan plots by ranking the ccFDR for each SNPs.
Function annotation of pleiotropic SNPs
The GOEAST (http://omicslab.genetics.ac.cn/GOEAST/), a web based software toolkit, was applied to identify significantly enriched gene ontology terms among a given list of novel SNPs annotated genes. The p-values were computed by hyper-geometric tests and adjusted for multiple comparisons by stringent Yekutieli (FDR under dependency) method [21]. In order to partially explore and visualize the functional partnership and interaction of the identified genes for T2D and CBMI, the corresponding protein association networks were constructed using the STRING 10.0 database (http://string-db.org/).
In order to characterize the functional role of identified pleiotropic SNPs for T2D and CO, we annotated each pleiotropic SNP to corresponding known or predicted regulatory regions (e.g., DNAase hypersensitivity) in the intergenic regions of the human genome using RegulomeDB (Version 1.1, http://www.regulomedb.org/index), Kyoto Encyclopedia of Genes and Genomes (KEGG) (https://www.genome.jp/kegg/), GeneCards (https://www.genecards.org/), and HaploReg (Version 4.1, http://www.broadinstitute.org/mammals/haploreg/haploreg.php) tools. RegulomeDB retrieves the functional annotation from ENCODE/GEO database and calculates a score for the regional regulatory potential. HaploReg visualizes the linked interesting SNPs and small indels to chromatin state and protein binding; the user can configure parameters (such as the LD threshold and the reference population used) from 1000 genomes project.
Results
Pleiotropic enrichment and genetic correlation of T2D SNPs and CBMI SNPs
We applied conditional Q–Q plot to graphically test pleiotropic enrichment of genetic loci. A higher proportion of true associations was demonstrated as earlier shift to left. A greater spacing between each curve intuitively shows a stronger pleiotropic enrichment shared by both traits. As shown in Fig. 1a, a great separation between each curve was observed, which indicates a strong enrichment of T2D-associated SNPs. The proportion of true association in T2D varies across different levels of effects for CBMI. We also observed a moderate enrichment for CBMI conditioned on T2D (Fig. 1b) contrast with the profile for T2D conditioned on CBMI. According to the linkage disequilibrium score regression (LDSC), we also found that there was genetic correlation between T2D and CO (genetic correlation = 0.21, p = 0.002).

Conditional Q–Q plot. Stratified Q–Q plots of enrichment vs. nominal −log10(p-values) (corrected for inflation) in a T2D below the standard GWAS threshold of p < 5 × 10−8 as a function of significance of the association with CBMI at the level of −log10(p) > 0, −log10(p) > 1, −log10(p) > 2, −log10(p) > 3 corresponding to p < 1, p < 0.1, p < 0.01, p < 0.001, respectively, and in b CBMI below the standard GWAS threshold of p < 5 × 10−8 as a function of significance of the association with T2D at the level of −log10(p) > 0, −log10(p) > 1, −log10(p) > 2, −log10(p) > 3 corresponding to p < 1, p < 0.1, p < 0.01, p < 0.001, respectively. Dashed lines indicate the null-hypothesis
T2D loci identified by cFDR
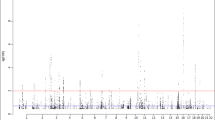
We constructed a cFDR Manhattan plot to illustrate the various chromosomal locations of significant SNPs, which are associated with T2D conditioned on CBMI (Fig. S1). We identified 139 significant SNPs with a cFDR significance threshold <0.05 (Table S1), and 14 of these SNPs reached to genome-wide significance threshold (p < 5 × 10-8) in the original meta-analysis for T2D [14]. Importantly, we identified 125 novel SNPs missed in the original study. By applying a strict conservative threshold with cFDR <0.01, 59 loci remained (Table S1). Notably five SNPs, including three novel SNPs, rs231354 (cFDR = 6.80E-04), rs3852527 (cFDR = 4.30E-03), and rs234857 (cFDR = 1.19E-02), were mapped to the gene KCNQ1 (11p15.4). This gene has been reported to be associated with increased susceptibility to T2D in previous studies [22, 23].
In order to partially test and characterize the functional partnership in the identified T2D target genes and their biological interaction networks, the 139 significant SNPs annotated genes were scanned by the STRING 10.0. Notably, the network consisted of genes that play a key role in multicellular organismal development, anatomical structure development and positive regulation of cellular component organization. These results revealed significant association between the topological features and biological function of T2D target genes (Fig. 2a). To systematically investigate the functional enrichment of the observed novel SNPs target genes, we conduct gene ontology analysis using GOEAST (Table 1). Interestingly, the results showed a cluster of biological processes in central nervous system, e.g., regulation of glial cell differentiation (p = 3.67E-18), regulation of neurogenesis (p = 1.73E-11), and central nervous system neuron differentiation (p = 4.65E-10), which may play a potential function in the pathogenesis of T2D.

A functional protein association network analysis for T2D (a) and CBMI (b) susceptibility genes. Connections are based on co-expression and experimental evidence with a STRING 10.0 summary score above 0.4. Each filled node denotes a gene; edges between nodes indicate protein–protein interactions between protein products of the corresponding genes. Different edge colors represent the types of evidence for the association
CBMI loci identified by cFDR
As illustrated in the cFDR Manhattan plot for CBMI (Fig. S2), we identified a total of 37 significant SNPs including 31 novel loci (cFDR < 0.05) for CBMI variation based on T2D (Table S2). These loci were mapped to 17 different chromosomes. Interestingly, the novel SNP rs934778 located at gene POMC has been reported to be associated with T2D earlier [24]. By applying a strict conservative threshold with cFDR <0.01, 25 loci remained. More importantly, the current method validated six loci (rs4883723, rs1344840, rs2635727, rs1031477, rs7553348, and rs17024393), which were identified in the original CBMI GWAS study [15]. Functional interaction network analysis among the 37 significant SNPs corresponding genes showed a weaker gene–gene interaction (Fig. 2b) compared with T2D SNPs corresponding genes, gene enrichment analysis shows that the network consisted of genes, which play an important role in cyclic nucleotide biosynthetic process, sensory organ development and intracellular signal transduction (Table S3).
Pleiotropic loci for T2D and CBMI
In order to test the SNPs that were associated with both T2D and CBMI, we computed ccFDR and constructed a ccFDR Manhattan plot (Fig. 3). Ten novel pleiotropic SNPs mapped to eight regions, which reached a significance level of ccFDR < 0.05 (Table 2). These 10 loci were annotated to 16 different genes, of which 15 (LOC100133285, LOC387930, KIAA1602, GNAT2, GNPDA2, LOC100131309, MFAP3, GALNT10, MTCO1P2, NPAS3, LOC400652, LOC342784, LOC390415, LOC100128339, PDILT, and FRAP1) were novel ones, and gene GNPDA2 was previously reported in both T2D and CBMI GWASs [7].

Conjunctional Manhattan plot for T2D and CBMI. SNPs with conjunctional-log10(cFDR) > 1.3 (i.e., cFDR < 0.05) are shown above the red line. The figure marks the chromosomal locations of significant loci. Details about all significant loci are offered in Table 2 (color figure online)
We performed a series of functional analyses to explore the potential regulatory function for ten pleiotropic SNPs. By using RegulomeDB and HaploReg databases, rs1031477 (NCKAP5L), rs649721 (LOC400652), rs17024393 (GNAT2), rs4883723 (LOC387930), rs1441264 (LOC100128339), and rs1996023 (GNPDA2) overlapped with open chromatin in different cell lines, e.g., monocyte CD14 + and fibroblasts, which are potentially related to the pathogenesis of T2D and/or CO. Furthermore, the DNA regions containing rs17024393 (GNAT2), rs649721 (LOC400652), and rs7715256 (MFAP3) overlapped with enhancer regions in several tissues/cell lines, such as primary mononuclear cells, heart and muscle etc., confirming that our pleiotropic SNPs are strongly clustered in regions with active chromatin state. By using KEGG database, we found that rs1996023 (GNPDA2) was in the metabolic pathway, especially related to amino sugar and nucleotide sugar metabolism. By investigating the GeneCards annotation results (https://www.genecards.org/), we observed that FRAP1 was a central regulator of cellular metabolism, growth, and survival in response to hormones, growth factors, nutrients, energy, and stress signals.
Discussion
Here, by applying the cFDR method to two independent GWASs with summary statistics of T2D and CBMI, we identified 139 T2D susceptibility SNPs including 125 novel SNPs, which were overlooked in the original study [14]. Furthermore, we identified 37 SNPs located in 17 chromosomes were significantly associated with CBMI conditioned on T2D (cFDR < 0.05). Importantly, there were ten pleiotropic SNPs, which suggested a shared genetic mechanism among T2D and CBMI. Our results demonstrated that GWAS from CBMI can enhance the identification of T2D susceptibility loci and improve our understanding of the influence of pleiotropic on both phenotypes.
The cFDR method shows that rs9366354 is most significant novel T2D susceptibility SNP. It was mapped to the intron of gene CDKAL1 (6p22), CDKAL1 is a member of the methylthiotransferase family and it plays a key role in transferase activity and iron-sulfur cluster binding. CDKAL1 has been reported to be associated with insulin resistance, which is a clinical marker related to T2D [25]. There is an interesting SNP rs3760511 located at the gene HNF1B (17q12). This gene encodes a homeodomain-containing transcription factor and it is a critical regulator for the differentiation in the embryonic pancreas development [26,27,28]. Mutations in HNF1B result in maturity-onset diabetes of the young type 5 (MODY5) [29] and early-onset T2D [30]. Previous study reported that this gene can induce abnormal glucose tolerance and reduced insulin sensitivity, it may cause the enhancement of insulin secretion and the activation of the IGF1 pathway associated with the pathogenesis of T2D [31]. In addition, Kornfeld et al. [32] found that direct silencing of HNF1B results from obesity-induced overexpression of miR-802, which may cause glucose intolerance, impaired insulin signaling, and stimulated gluconeogenesis in the liver and suggested a critical role for HNF1B in the process of developing hepatic insulin resistance and modulating glucose metabolism. Another interesting SNP rs1326941 located in the APIP gene (11p13), although APIP was not reported to be associated with the susceptibility of T2D. However, the gene APIP involves in many physiological and pathophysiological activities through affecting ERK1/2 signaling pathway activation [33, 34]. For example, ERK1/2 signaling pathway has an important influence on the pathogenesis of T2D by controlling phosphorylation amount of cAMP-responsive element-binding protein, which plays an important role in glucose-mediated pancreatic beta-cell survival [34].
In addition, the present cFDR approach observed 37 SNPs for CBMI conditioned on T2D (cFDR < 0.05), and 6 SNPs were reported to be associated with CBMI in the original CBMI GWAS study [15]. We discovered 31 new loci, e.g., the novel significant SNP rs934778 was annotated to gene POMC. This gene is a precursor polypeptide hormone, the brainstem nucleus neurons and the pituitary gland etc. [35]. It is reported that POMC-expressing neurons has an important role in the maintenance of homeostatic mechanisms [36, 37]. Recent study showed that gene POMC, as a possible additional link, play an important role between T2D and central obesity [24].
Importantly, we identified ten novel pleiotropic loci (rs4883723 in LOC100133285 (LOC387930), rs1031477 in KIAA1602, rs17024393 in GNAT2, rs1996023 in GNPDA2 (LOC100131309), rs7715256 in MFAP3 (GALNT10), rs17522122 in MTCO1P2 (NPAS3), rs649721 in LOC400652 (LOC342784), rs1441264 in LOC390415 (LOC100128339), rs11864909 in PDILT, and rs1010447 in FRAP1) that have not been reported in previous publications in association with T2D or CO. GNPDA2 increases the risk of susceptibility for T2D and CO through the central nervous system processes in weight regulation [7]. The central nervous system has several possible roles in the regulation of body weight, energy metabolism, and other behaviors [38].
We should also notice that the heritability should be verified in different population [39]. Therefore, we can repeat the analysis in an independent population. If the validation failed, it might be caused by genetic diversity. In this study, we chose the largest sample size of GWASs in the research field, and other populations (such as Hispanics, Africans) can also be applied to this work in the future. The inter-ethnic study can provide an important information about relationship between T2D and CO among different populations.
The major advance of this study is that we enhanced the identification of novel T2D SNPs and pleiotropic SNPs by integrating GWAS summary statistics from T2D and CO phenotypes. Our results not only present novel insights for exploring common molecular mechanisms underlying T2D and CBMI, but also provide promising candidates for further experimental validation. Results presented, however, may have some limitations. First, we cannot associate the genetic variation with outcomes as our inability to access the clinical and genotype data. Whereas, the main objective of our study is to enhance the detection of disease associated genes and identify the common biological mechanisms in the two traits. Second, we only identified partial of previously reported genes in T2D and CO, as current study used a subset of the public available meta-analysis to reveal the missing heritability in T2D and CO. Therefore, further replication analysis and functional biological experiments are necessary to confirm our results. SNPs within a gene can affect other genes, which limited our precise gene annotation. In this study, we aim to find a potential gene for functional discussion, we chose the corresponded or nearest one. But in the future, we can integrate other information, such as expression quantitative trait loci (eQTL) data to get a more accurate gene annotation [40].
In conclusion, we illustrated the enhanced efficiency of the cFDR approach in identification of novel SNPs for T2D and CO. Our results provide an insight into the common genetic mechanisms in two traits, which provide prominent loci for further functional biological experiments and clinical replication.
References
American Diabetes Association. Classification and diagnosis of diabetes. Diabetes Care. 2016;39:S13–22. http://care.diabetesjournals.org/cgi/doi/10.2337/dc15-S005
Saddik B, Al-Dulaijan N. Diabetic patients’ willingness to use tele-technology to manage their disease. Online J Public Health Inform. 2015;7. http://journals.uic.edu/ojs/index.php/ojphi/article/view/6011
Lu Y, Loos RJF. Obesity genomics: Assessing the transferability of susceptibility loci across diverse populations. Genome Med. 2013;5:1.
Wright CM, Parker L, Lamont D, et al. Implications of childhood obesity for adult health: findings from thousand families cohort study. Brit Med J. 2001;323:1280–4.
Twig G, Tirosh A, Leiba A, et al. BMI at age 17 years and diabetes mortality in midlife: A nationwide cohort of 2.3 million adolescents. Diabetes Care. 2016;39:1996–2003.
Llewellyn A, Simmonds M, Owen CG, et al. Childhood obesity as a predictor of morbidity in adulthood: A systematic review and meta-analysis. Obes Rev. 2016;17:56–67.
Willer CJ, Speliotes EK, Loos RJF, et al. Six new loci associated with body mass index highlight a neuronal influence on body weight regulation. Nat Genet. 2009;41:25–34. http://www.nature.com/doifinder/10.1038/ng.287
Ali O. Genetics of type 2 diabetes. World J Diabetes. 2013;4:114 http://www.wjgnet.com/1948-9358/full/v4/i4/114.htm
Yang J, Manolio TA, Pasquale LR, et al. Genome partitioning of genetic variation for complex traits using common SNPs. Nat Genet. 2011;43:519–25.
Dering C, Hemmelmann C, Pugh E, et al. Statistical analysis of rare sequence variants: An overview of collapsing methods. Genet Epidemiol. 2011;35:S12–S17.
Golan D, Lander ES, Rosset S. Measuring missing heritability: Inferring the contribution of common variants. Proc Natl Acad Sci. 2014;111:E5272–81. http://www.pnas.org/lookup/doi/10.1073/pnas.1419064111
Desikan RS, Schork AJ, Wang Y, Thompson WK, Dehghan A, Ridker PM et al. Polygenic overlap between C-reactive protein, plasma lipids and Alzheimer disease. Circulation. 2015;131:2061–69.
Desikan RS, Schork AJ, Wang Y, Witoelar A, Sharma M, McEvoy LK et al. Genetic overlap between Alzheimer's disease and Parkinson’s disease at the MAPT locus. Mol Psychiatry. 2015;20:1588–95.
Morris A, Voight B, Teslovich T. Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat Genet. 2012;44:981–90. http://test.europepmc.org/articles/PMC3442244
Felix JF, Bradfield JP, Monnereau C, et al. Genome-wide association analysis identifies three new susceptibility loci for childhood body mass index. Hum Mol Genet. 2016;25:389–403.
Zeng CP, Chen YC, Lin X, et al. Increased identification of novel variants in type 2 diabetes, birth weight and their pleiotropic loci. J Diabetes. 2017;9:898–907.
Price AL, Patterson NJ, Plenge RM, et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904–9.
Bulik-Sullivan BK, Loh PR, Finucane HK, et al. LD score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat Genet. 2015;47:291.
Bulik-Sullivan B, Finucane HK, Anttila V, et al. An atlas of genetic correlations across human diseases and traits. Nat Genet 2015;47:1236.
Liley J, Wallace C. A pleiotropy-informed Bayesian false discovery rate adapted to a shared control design finds new disease associations from GWAS summary statistics. PLoS Genet. 2015;11:e1004926.
Yekutieli D, Benjamini Y. Resampling-based false discovery rate controlling multiple test procedures for correlated test statistics. J Stat Plan Inference. 1999;82:171–96. http://linkinghub.elsevier.com/retrieve/pii/S0378375899000415
Unoki H, Takahashi A, Kawaguchi T, et al. SNPs in KCNQ1 are associated with susceptibility to type 2 diabetes in East Asian and European populations. Nat Genet. 2008;40:1098–102.
Hanson RL, Guo T, Muller YL, et al. Strong parent-of-Origin effects in the association of kcnq1 variants with type 2 diabetes in american indians. Diabetes. 2013;62:2984–91.
Mencarelli M, Zulian A, Cancello R, et al. A novel missense mutation in the signal peptide of the human POMC gene: A possible additional link between early-onset type 2 diabetes and obesity. Eur J Hum Genet. 2012;20:1290–4.
Billings LK, Florez JC. The genetics of type 2 diabetes: What have we learned from GWAS? Ann N Y Acad Sci. 2010;1212:59–77.
Coffinier C, Thepot D, Babinet C, et al. Essential role for the homeoprotein vHNF1/HNF1beta in visceral endoderm differentiation. Development. 1999;126:4785–94. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=10518495
Maestro MA, Boj SF, Luco RF, et al. Hnf6 and Tcf2 (MODY5) are linked in a gene network operating in a precursor cell domain of the embryonic pancreas. Hum Mol Genet. 2003;12:3307–14.
Servitja JM, Ferrer J. Transcriptional networks controlling pancreatic development and beta cell function. Diabetologia. 2004;47:597–613.
Horikawa Y, Iwasaki N, Hara M, et al. Mutation in hepatocyte nuclear factor-1ß gene (TCF2) associated with MODY. Nat Genet. 1997;17:384–5.
Gupta V, Vinay DG, Rafiq S, et al. Association analysis of 31 common polymorphisms with type 2 diabetes and its related traits in Indian sib pairs. Diabetologia. 2012;55:349–57.
Ríos-Tamayo R, Lupiañez CB, Campa D, et al. A common variant within the HNF1B gene is associated with overall survival of multiple myeloma patients: results from the IMMEnSE consortium and meta-analysis. Oncotarget. 2016;7:59029–48. http://www.oncotarget.com/fulltext/10665
Kornfeld JW, Baitzel C, Könner AC, et al. Obesity-induced overexpression of miR-802 impairs glucose metabolism through silencing of Hnf1b. Nature. 2013;494:111–5.
Hong S, Lee WJ, Kim YD, et al. APIP, an ERBB3-binding partner, stimulates erbB2-3 heterodimer formation to promote tumorigenesis. Oncotarget. 2016;7. http://www.ncbi.nlm.nih.gov/pubmed/26942872.
Costes S, Broca C, Bertrand G, et al. ERK1/2 control phosphorylation and protein level of cAMP-responsive element-binding protein: A key role in glucose-mediated pancreatic β-cell survival. Diabetes. 2006;55:2220–30.
Mountjoy KG. Functions for pro-opiomelanocortin-derived peptides in obesity and diabetes 58. BiochemJ. 2010;428:305–24.
Parton LE, Ye CP, Coppari R, et al. Glucose sensing by POMC neurons regulates glucose homeostasis and is impaired in obesity. Nature. 2007;449:228–32.
Fick LJ, Belsham DD. Nutrient sensing and insulin signaling in neuropeptide-expressing immortalized, hypothalamic neurons: A cellular model of insulin resistance. Cell Cycle. 2010;9:3186–93.
Berthoud H-R, Berthoud H-R, Morrison C, et al. The brain, appetite, and obesity. Annu Rev Psychol. 2008;59:55–92. http://www.ncbi.nlm.nih.gov/pubmed/18154499
Musani SK, Martin LJ, Woo JG, et al. Heritability of the severity of the metabolic syndrome in whites and blacks in 3 large cohorts. Circ Cardiovasc Genet. 2017;10:e001621.
Thibodeau SN, French AJ, McDonnell SK, et al. Identification of candidate genes for prostate cancer-risk SNPs utilizing a normal prostate tissue eQTL data set. Nat Commun. 2015;6:8653.
Acknowledgements
This study was partially funded by grants from the National Institutes of Health (U19AG055373, P50AR055081, R01AR057049, R01AR059781, D43TW009107, P20GM109036, R01MH107354, R01MH104680, R01GM109068), the Edward G. Schlieder Endowment fund to Tulane University, the Medical research fund of Guangdong province (A2017575). We thank the DIAGRAM Consortium and the Early Growth Genetics Consortium for the summary statistics of GWASs data.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethics statement
This paper re-analyses previously published datasets. The relevant institutional review boards or ethics committees of the participating organizations approved the research protocol of the individual GWAS used in the current analysis.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Zeng, CP., Lin, X., Peng, C. et al. Identification of novel genetic variants for type 2 diabetes, childhood obesity, and their pleiotropic loci. J Hum Genet 64, 369–377 (2019). https://doi.org/10.1038/s10038-019-0577-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s10038-019-0577-5
This article is cited by
-
The Potential of Albuminuria as a Biomarker of Diabetic Complications
Cardiovascular Drugs and Therapy (2021)
