
Abstract
Carrier screening of spinal muscular atrophy (SMA) can provide reproductive options for carriers and prevent the birth defects. Here, we developed a simple screening test based on melting analysis. The test comprises a duplex PCR with two primer pairs and three probes to simultaneous amplify SMN1, SMN2, and CFTR. By analyzing the melting profiles, we were able to determine the SMN1/SMN2 ratio and SMN1 + SMN2 copy number to subsequently determine the copy number of SMN1. Samples with one copy of SMN1 were considered as “high risk for carrier,” while samples with ≥2 copies of SMN1 were considered as “low risk for carrier.” We evaluated the clinical performance of this test using 215 clinical samples with various genotypes that had been previously confirmed by multiplex ligation-dependent probe amplification (MLPA). The test showed high sensitivity (100%) and specificity (97.1%) as well as high positive (97.3%) and negative (100%) predictive value, and was in perfect agreement with the gold standard test, MLPA (k = 0.97). Moreover, it is rapid, inexpensive, and easy to perform and automate, with high reproducibility and capacity. Therefore, we expect this test will advance carrier screening for SMA.
Similar content being viewed by others

Introduction
Spinal muscular atrophy (SMA) is an autosomal recessive disease and is one of the most common genetic causes of infant death worldwide [1]. Among different populations, the carrier frequency varies from 1:47 to 1:72, with an estimated incidence of 1 in 6000–10000 live births [2].
Patients with SMA exhibit a continuous range of severity, and the disease is generally classified into one of four clinical types based on the age of onset and phenotype: type I (onset before 6 months of age with no ability to sit unaided), type II (onset before 18 months of age with the ability to sit but not walk independently), type III (onset after 18 months of age with the ability to walk), and type IV (adult onset with mild symptoms) [3, 4]. All types of SMA are caused by homozygous defects in the survival motor neuron 1 gene, SMN1, which is located on 5q 13.2. The product of SMN1, survival motor neuron (SMN) protein, is critical for maintaining the physical function of skeletal and intercostal muscles [5]. Approximately 95% of SMA patients lose both alleles of SMN1, while the rest may carry a deletion of one allele and an intragenic mutation on the other [6].
Survival motor neuron 2 (SMN2), which is highly similar to SMN1, is located in the same chromosome region as SMN1. SMN1 and SMN2 differ at just five nucleotides; four of which are located in non-coding regions [5]. Although the exclusive difference in the coding region (c. 840 C>T) in exon 7 of SMN2 does not alter the amino acid sequence, it causes exon skipping, and as a result, 50–90% of the protein products are improperly spliced [7]. However, SMN2 does produce a limited amount of functional, full-length SMN protein, which can partially rescue the phenotype caused by a defect in SMN1. Therefore, the clinical manifestation in SMA patients can be alleviated by an increase in the SMN2 copy number [8].
Considering the potentially severe phenotype and the high prevalence of SMA, carrier screening programs (in either the pre-conception or prenatal period) are acknowledged to be beneficial, as they can provide reproductive options for carriers and prevent the birth defects, although whether the general population or just a specific population should be offered screening is still controversial [9,10,11]. In addition to the phenotype and prevalence, the availability of technically and clinically valid screening methods, with high capacity and reasonable cost, is also an important factor to be considered when implementing a carrier screening program for SMA [11].
Based on the technical standards and guidelines for SMA testing, current carrier screening relies on an accurate determination of the SMN1 copy number [12]. Generally, carriers have one copy of SMN1 and noncarriers have two or, occasionally, more copies of SMN1. In this study, we introduced a simple test based on melting analysis to determine the copy number of SMN1, and we evaluated its clinical performance using 215 clinical samples. We aimed to provide an alternative method based on the widespread qPCR platform to advance the implementation of a carrier screening program for SMA.
Materials and methods
Design of the screening test
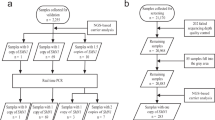
The key components of the test were two primer pairs and three molecular beacon probes (Fig. 1a). One primer pair (F1 and R1) targets the identical regions in SMN1 and SMN2, and the other primer pair (F2 and R2) targets a conserved region in CFTR. A ROX-labeled probe (P1) was designed as a perfect match to SMN1 but a mismatch to SMN2 at the c. 840 nucleotide. During melting, P1 is able to hybridize to the target sequences amplified from both SMN1 and SMN2, generating two distinct peaks in the resulting melting profiles in the ROX channel (Fig. 1b). A FAM-labeled probe (P2) targets the identical regions in SMN1 and SMN2, and another FAM-labeled probe (P3) targets CFTR. During melting, P2 and P3 generate two distinct peaks in the resulting melting profiles in the FAM channel (Fig. 1b). The sequences of the primers and probes are available in Supplemental Table 1.

Design of the screening test. a Primers and probes in the duplex PCR. b Typical melting profiles derived from the duplex PCR. c Determination of SMN1 copy number with SMN1/SMN2 ratio and the copy number of SMN1+SMN2. d Algorithm for normalized relative peak value
Theoretically, the ratio of SMN1/SMN2 can be determined via the relative area of the corresponding melting peaks in the ROX channel. Similarly, the ratio of (SMN1 + SMN2)/CFTR can be evaluated in the FAM channel, and the copy number of SMN1 + SMN2 is calculable since CFTR is constant in the human genome, at two copies. Through combined analysis of the SMN1/SMN2 ratio and the copy number of SMN1 + SMN2, we can evaluate the copy number of SMN1, and thus screen for carriers (Fig. 1c). However, it is difficult to accurately quantify the peak area with most qPCR software. Therefore, we used peak height instead of peak area to examine the relative copy numbers for the sake of simplification and compatibility (Fig. 1b). To reduce variance between tests and equipment, we used a mixed sample containing two copies of both SMN1 and SMN2 as a reference to introduce a normalized relative peak (NRP) algorithm for the measurement of relative copy numbers (Fig. 1d) [13].
The flowchart of the screening test is shown in Fig. 2. The test begins with the duplex PCR. After amplification, the PCR products are subjected to melting analysis. We first analyzed the ROX channel in which three melting profiles could be observed, the SMN2-derived single peak, SMN1-derived single peak, and SMN1 and SMN2-derived double peaks. Samples with an SMN2-derived single peak are considered to be lacking SMN1, and are classified as “high risk for SMA”; samples with an SMN1-derived single peak are classified as “uncertain”; and samples with double peaks are classified as “high risk for carrier”, “low risk for carrier”, and “uncertain” based on the NRP values. Next, samples considered as uncertain from the ROX channel analysis were further classified based on their NRP values in the FAM channel. Eventually, all of the samples can be classified as “high risk for carrier” or “low risk for carrier” through combined analysis of their melting profiles in the two fluorescent channels.

Flowchart of the screening test
Reproducibility of the screening test
Sixteen carrier samples with one copy of SMN1 and two copies of SMN2 (1 SMN1/2 SMN2), 15 noncarrier samples with two copies of both SMN1 and SMN2 (2 SMN1/2 SMN2), and a reference sample were examined with the screening test on three consecutive days. The NRP values of these characteristic genotypes were analyzed to evaluate the reproducibility of the screening test.
Cut-off value for the screening test
To define cut-off values in the ROX channel for samples containing both SMN1 and SMN2, 30 samples, including six genotypes with different SMN1/SMN2 ratios, were examined with the screening test on three consecutive days. The NRP values in the ROX channel collected on the three days were combined to obtain genotype-specific NRP ranges, and cut-off values were defined based on the NRP range of each specific genotype. Similarly, for samples containing both SMN1 and SMN2, the cut-off value in the FAM channel was defined with 36 samples, including three genotypes with specific SMN1 + SMN2 copy numbers. In samples containing SMN1 alone, the cut-off value in the FAM channel was defined with 17 samples, including two genotypes with different copy numbers of SMN1.
Clinical performance
A blinded test was performed using 215 clinical samples whose genotypes had been previously validated with multiplex ligation-dependent probe amplification (MLPA) [14]. The genotype information for these samples was withheld from the technician who performed the screening tests, and the screening results were compared to those from MLPA by another technician to evaluate the clinical performance of the screening test.
Samples
The blood and DNA samples used in this study were collected from the molecular diagnostics laboratory of Xiamen Maternal and Child Health Hospital. For EDTA-anticoagulated whole blood samples, genomic DNA was extracted with the QIAamp DNA mini Kit (Qiagen, Valencia, CA, USA) according to the manufacturer’s protocol. The DNA concentration was determined by measuring the absorbance at 260 nm using a NanoDrop 2000 spectrophotometer (Thermo Fisher, Waltham, MA, USA).
A total of 50 DNA samples, which had been confirmed to contain two copies of both SMN1 and SMN2, were diluted to 10 ng/μL and mixed in equal amounts to prepare a reference DNA sample that was used to normalize relative peak values.
PCR and melting analysis
PCR and melting analysis were performed on a CFX 96 thermocycler (Bio-Rad, Hercules, CA, USA). Each 25 μL reaction contained 180 nmol/L F1, 18 nmol/L R1, 16 nmol/L F2, 160 nmol/L R2, 32 nmol/L P1, 40 nmol/L P2, 40 nmol/L P3, 67 mmol/L Tris-HCl (pH 8.6), 16 mmol/L (NH4)2SO4, 0.01% Tween 20, 1 U of TaqHS (Takara), 4 mmol/L Mg2+, 0.2 mmol/L each dNTP, and 50 ng of template DNA.
The cycling conditions were as follows: 95 °C for 5 min, followed by 40 cycles of 95 °C for 20 s, 57 °C for 20 s, and 72 °C for 20 s. The melting analysis began with denaturation at 95 °C for 1 min and renaturation at 40 °C for 2 min, followed by melting from 50 °C to 75 °C, with a ramp rate of 0.1 °C/s. Fluorescence signals were collected every 0.5 °C.
MLPA
The MLPA assay was performed and analyzed according to the manufacturer’s instructions of the SALSA® MLPA kit (Catalogue number: P060, MRC-Holland, Amsterdam, the Netherlands). SALSA® Reference DNA (Catalogue number: SD019, MRC-Holland), which contains two copies of both SMN1 and SMN2, was used as normal control. The amplification products of DNA samples were electrophoresed on an ABI 3130 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA) and the SMN1 and SMN2 copy numbers were analyzed with the Coffalyzer software (MRC-Holland). In brief, the SMN1 and SMN2 copy numbers were evaluated according to the range of dosage quotient (DQ) values: DQ values between 0.4–0.65, 0.8–1.2, 1.3–1.65, and 1.75–2.15 indicates the presence of one, two, three, and four copies of corresponding gene, respectively.
Statistical analysis
Each sample was tested in triplicate, and the mean NRP value was used for analysis. A normality test was conducted to determine if a set of NRP values followed a normal distribution. When the NRP value data set was non-normally distributed, a non-parametric method, Kruskal–Wallis analysis of variance (ANOVA) [15], was used to test whether samples originated from the same distribution. All statistical analyses were performed using OriginPro 8.0 software (OriginLab Corp., Northampton, MA, USA).
Ethics statement
The samples used in this study were remainders from previous tests, and no additional sampling was performed. Except for the MLPA results, identifying information, including the names and ages of the patients, were withheld from the study group. Therefore, no written informed consent was required. The study protocol was approved by the Research Ethics Committees of Xiamen Maternal and Child Health Hospital.
Results
Reproducibility of the screening test
The NRP values of both genotypes in both channels were not normally distributed. Therefore, Kruskal–Wallis ANOVA was used to evaluate the reproducibility of the test. As shown in Fig. 3, the NRP values among the three testing days were regarded as originating from the same distribution (P > 0.05) for both the carrier and noncarrier sample groups in both fluorescent channels. Therefore, the screening test showed high reproducibility.

Reproducibility of the screening test. NRP values obtained on three consecutive days were compared with the Kruskal–Wallis ANOVA for carrier samples (a) and noncarrier samples (b). Statistical analysis: the line within the box denotes the median, the square within the box denotes the mean, the horizontal borders of each box denote the 25th and 75th percentiles, the whiskers denote the 5th and 95th percentiles, and the stars denote the maximum and minimum
Cut-off values for the screening test
To define cut-off values for samples containing both SMN1 and SMN2, six genotypes with different SMN1/SMN2 ratios were examined by the screening test, which displayed different melting profiles in the ROX channel (Supplemental Fig. 1A), and their NRP ranges were plotted (Fig. 4a). When the SMN1/SMN2 ≤ 1/2, the samples are highly likely to contain one copy of SMN1, and thus were considered as “high risk for carrier.” When the SMN1/SMN2 ≥ 2/1, the samples are highly likely to contain more than one copy of SMN1, and thus were considered as “low risk for carrier.” When the SMN1/SMN2 = 1 (e.g., 1/1 or 2/2), it was difficult to directly measure the SMN1 copy number; thus, these samples were considered as “uncertain.” For samples in which SMN1/SMN2 = 2/3, combined analysis with the FAM channel proved to be beneficial to the accuracy in our preliminary study. Therefore, these samples were considered to be “uncertain” as well. Based on the above, we defined mean values for adjacent maximum and minimum between classifications as the cut-off values. Finally, 0.6 and 1.27 were defined as the cut-off values in the ROX channel for classifying “high risk,” “low risk,” and “uncertain” samples (Fig. 4a). Three “uncertain” genotypes with different SMN1 + SMN2 copy numbers were examined in the FAM channel to evaluate their NRP ranges (Supplemental Fig. 1B). As shown in Fig. 4b, when SMN1 + SMN2 = 2, the samples are highly likely to contain one copy of SMN1, and thus were considered as “high risk for carrier.” When SMN1 + SMN2 ≥ 4, the samples are highly likely to contain two or more copies of SMN1, and thus were considered as “low risk for carrier.” Similarly, the mean NRP value (0.91) of the adjacent maximum and minimum between two and four copies of SMN1 + SMN2 was defined as the cut-off value in the FAM channel for classifying “high risk” and “low risk” samples.

Cut-off values of the screening test. Determination of cut-off values in ROX channel (a) and FAM channel (b) for samples contain both SMN1 and SMN2. c Determination of cut-off value in FAM channel for samples contain SMN1 alone. Statistical analysis: the line within the box denotes the median, the square within the box denotes the mean, the horizontal borders of each box denote the 25th and 75th percentiles, the whiskers denote the 5th and 95th percentiles, and the stars denote the maximum and minimum
To define a cut-off value for samples containing SMN1 alone, two genotypes with different SMN1 copy numbers were examined by the screening test, which displayed different melting profiles (Supplemental Fig. 1C and D), and their NRP ranges in the FAM channel were plotted (Fig. 4c). When the copy number of SMN1 was one, the samples were considered as “high risk for carrier.” When the copy number of SMN1 ≥ 2, the samples were considered as “low risk for carrier.” Therefore, the mean NRP value (0.64) of the maximum and minimum between samples with one and two copies of SMN1 was defined as the cut-off value for classifying “high risk” and “low risk” samples.
Clinical performance
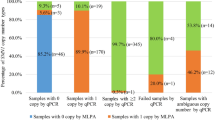
Among the melting profiles of the 215 clinical samples in the ROX channel, six displayed SMN1-derived single peak and 209 samples displayed double peaks. According to the cut-off values, 94, 45, and 70 of the clinical samples with double peaks were classified as “high risk for carrier,” “low risk for carrier,” and “uncertain,” respectively (Fig. 5a). Six samples containing single peaks and 70 uncertain samples containing double peaks were further analyzed in the FAM channel. Of the samples containing single peaks, one and five samples were classified as “high risk for carrier” and “low risk for carrier,” respectively (Fig. 5b). Of the samples containing double peaks, 18 and 52 samples were classified as “high risk for carrier” and “low risk for carrier,” respectively (Fig. 5c). Finally, 113 and 102 samples were classified as “high risk for carrier” and “low risk for carrier,” respectively (Table 1).

Clinical validation of the screening test. a According to the defined cut-off values, samples contain both SMN1 and SMN2 were classified into three groups in ROX channel. Six samples containing single peaks (b) and 70 uncertain samples containing double peaks (c) were further analyzed in the FAM channel. The samples in red indicate the false positive samples
Comparison to the MLPA results showed that all of the carriers were detected by the screening test, indicating 100% sensitivity. Three samples containing two copies of both SMN1 and SMN2 were misclassified as “high risk for carrier” in the screening test, indicating 97.1% specificity. The positive and negative predictive values of the screening test were 97.3 and 100%, respectively. Cohen’s kappa coefficient (k) between the screening test and MLPA was 0.97.
Discussion
MLPA is generally acknowledged as a gold standard for determining SMA carriers due to its high degree of precision for the quantification of SMN1 copy number [14, 16,17,18]. Other platforms, such as next-generation sequencing (NGS) [19], digital PCR [20, 21], and liquid bead array [22, 23], have also been shown to be of high sensitivity and specificity for the detection of SMA carriers. However, in addition to being highly sensitive and specific, an ideal screening method should also be high capacity, rapid, inexpensive, and easy to perform and automate. Therefore, some issues remain to be resolved before these aforementioned platforms are able to be widely used for population-based screening. For example, MLPA requires a long turnaround time (~20 h), is relatively expensive, and is a multi-step method, including post-PCR capillary electrophoresis. Thus, it is more suitable for diagnostic use rather than screening. In addition to a long turnaround time, NGS also requires relative high sequencing depth to obtain sufficient carrier probability, which increases the costs. Moreover, the NGS platform is limited in underdeveloped regions. Similarly, digital PCR and liquid bead array is relatively costly and rarely used in underdeveloped regions. In contrast, qPCR is a basic platform that is widespread in clinical laboratories worldwide. It is simple, rapid, inexpensive, high capacity, and easy to perform and automate. Moreover, no post-PCR manipulations are required, thereby minimizing the risk of cross contamination. Therefore, qPCR would be an ideal platform for carrier screening of SMA.
The existing probe-based qPCR methods for SMA carrier screening rely on (1) absolute quantification of SMN1 copy number with an external reference [24] and (2) comparative quantification of SMN1 copy number with an internal reference [25]. Although the SMN2 copy number does not provide useful information for counseling in carrier screening, it influences the quantification of SMN1 due to the high similarity between SMN1 and SMN2. Therefore, accurate quantification of SMN1 requires effective elimination of the influence of SMN2. In absolute quantification method, SMN1 and SMN2 are amplified with a universal primer pair and quantified with different probes. In contrast, SMN1 and SMN2 are amplified with different primer pairs and quantified with a universal probe in comparative quantification method. In this regard, an absolute quantification method is preferred over a comparative quantification method because probes are more specific than primers for eliminating the influence of SMN2. However, absolute quantification methods require numerous external reference samples to establish a quantitative standard, which distinctly compromises test capacity. In contrast, a comparative quantification method only requires an external reference sample, thus maintaining high capacity. By exploiting the advantages of these methods and overcoming their limitations, we have developed an alternative screening method that relies on melting analysis, which combines high accuracy and high capacity. As shown in Fig. 4, the cut-off values were determined for different genotypes with various combinations of SMN1 and SMN2 copy numbers, while taking into consideration the contribution of SMN2, thus obtaining high accuracy (100% sensitivity and 97.1% specificity). Similar to a comparative quantification method, the intra-assay relative peak analysis in our method requires only one reference sample for normalization, thus maintaining high capacity. Moreover, when triplicate samples were assumed to be three independent samples and were re-evaluated in a clinical validation study, the performance of our test was also satisfactory (Supplemental Table 2). Therefore, replicates are optional, and the test capacity could reach a theoretical maximum of 95 samples per run in a 96-well qPCR thermocycler. High-resolution melting (HRM) is another qPCR-based method, which is promising for rapid carrier screening of SMA [4, 26,27,28]. However, HRM requires strict control of ramp rate which beyond the reach of some qPCR equipment. In this regard, our method is more compatible with the capacity of most qPCR equipment, suggesting a widespread use probability.
Three samples with two copies of both SMN1 and SMN2 were misclassified as “high risk for carrier” in the clinical validation study. A possible reason for this is the cut-off values, which were determined with limited numbers of samples, suggesting that further optimization is needed. In clinical practice, the cut-off values can be dynamically optimized according to a retrospective analysis with more samples of specific genotypes and the expected sensitivity and specificity. Theoretically, data analysis via peak area would be more accurate than peak height when accurate quantification of peak area is available. However, validation with the diagnostic method, e.g., MLPA or digital PCR, is required for “high risk carriers” whether the screening algorithm is based on peak height or area.
Hybrid SMN gene has been shown in a relatively high prevalence (~1/600) and could cause misclassification of SMN1 and SMN2 [21, 29]. However, this genetic variation does not affect clinical utility of our test due to the probe (P1) was designed to target the c.840 nucleotide, which is the key position for the biological function of SMN. It should, however, be noted that, similar to MLPA and most screening methods, some low-frequency carrier genotypes, e.g., those bearing two SMN1 genes in cis on a single chromosome or non-deletion mutations, would be missed by our method [12]. Therefore, the detection rate of the screening test would differ based on the prevalence of these carrier genotypes in different ethnic populations [30]. Moreover, in rare cases, mutations within the primer or probe binding sites, mosaicism, or other noncanonical SMN genes, could pose a finite risk to the accuracy of the screening test, thus complicate interpretation and carrier status calling [12].
In conclusion, we developed a simple test based on melting analysis on the qPCR platform and evaluated its clinical performance for carrier screening of SMA. The test was highly sensitive (100%) and specific (97.1%), and was in perfect agreement with the gold standard test, MLPA (k = 0.97). Moreover, it was rapid (2 h from DNA extraction to result export), inexpensive (0.6 USD/test), with high reproducibility and capacity, and easy to perform and automate. We expect this alternative test could advance carrier screening for SMA.
References
Minino AM, Xu J, Kochanek KD. Deaths: preliminary data for 2008. Natl Vital Stat Rep. 2010;59:1–52.
Sugarman EA, Nagan N, Zhu H, Akmaev VR, Zhou Z, Rohlfs EM, et al. Pan-ethnic carrier screening and prenatal diagnosis for spinal muscular atrophy: clinical laboratory analysis of >72,400 specimens. Eur J Hum Genet. 2012;20:27–32.
Munsat TL, Davies KE. International SMA consortium meeting. (26–28 June 1992, Bonn, Germany). Neuromuscul Disord. 1992;2:423–8.
Er TK, Kan TM, Su YF, Liu TC, Chang JG, Hung SY, et al. High-resolution melting (HRM) analysis as a feasible method for detecting spinal muscular atrophy via dried blood spots. Clin Chim Acta. 2012;413:1781–5.
Lefebvre S, Burglen L, Reboullet S, Clermont O, Burlet P, Viollet L, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell . 1995;80:155–65.
Wirth B. An update of the mutation spectrum of the survival motor neuron gene (SMN1) in autosomal recessive spinal muscular atrophy (SMA). Hum Mutat. 2000;15:228–37.
Kashima T, Manley JL. A negative element in SMN2 exon 7 inhibits splicing in spinal muscular atrophy. Nat Genet. 2003;34:460–3.
Gavrilov DK, Shi X, Das K, Gilliam TC, Wang CH. Differential SMN2 expression associated with SMA severity. Nat Genet. 1998;20:230–1.
Prior TW. Carrier screening for spinal muscular atrophy. Genet Med. 2008;10:840–2.
ACOG Committee on Genetics. ACOG committee opinion No. 432: spinal muscular atrophy. Obstet Gynecol. 2009;113:1194–6.
Muralidharan K, Wilson RB, Ogino S, Nagan N, Curtis C, Schrijver I. Population carrier screening for spinal muscular atrophy a position statement of the association for molecular pathology. J Mol Diagn. 2011;13:3–6.
Prior TW, Nagan N, Sugarman EA, Batish SD, Braastad C. Technical standards and guidelines for spinal muscular atrophy testing. Genet Med. 2011;13:686–94.
Zhou Y, Ge Y, Xiao L, Guo Q. Rapid and simultaneous screening of 47,XXY and AZF microdeletions by quadruplex real-time polymerase chain reaction. Reprod Biol. 2015;15:113–21.
Scarciolla O, Stuppia L, De Angelis MV, Murru S, Palka C, Giuliani R, et al. Spinal muscular atrophy genotyping by gene dosage using multiple ligation-dependent probe amplification. Neurogenetics . 2006;7:269–76.
Kruskal WH, Wallis WA. Use of ranks in one-criterion variance analysis. J Am Stat Assoc. 1952;47:583–621.
Arkblad EL, Darin N, Berg K, Kimber E, Brandberg G, Lindberg C, et al. Multiplex ligation-dependent probe amplification improves diagnostics in spinal muscular atrophy. Neuromuscul Disord. 2006;16:830–8.
Zhong Q, Bhattacharya S, Kotsopoulos S, Olson J, Taly V, Griffiths AD, et al. Multiplex digital PCR: breaking the one target per color barrier of quantitative PCR. Lab Chip. 2011;11:2167–74.
Stabley DL, Holbrook J, Harris AW, Swoboda KJ, Crawford TO, Sol-Church K, et al. Establishing a reference dataset for the authentication of spinal muscular atrophy cell lines using STR profiling and digital PCR. Neuromuscul Disord. 2017;27:439–46.
Larson JL, Silver AJ, Chan D, Borroto C, Spurrier B, Silver LM. Validation of a high resolution NGS method for detecting spinal muscular atrophy carriers among phase 3 participants in the 1000 Genomes Project. BMC Med Genet. 2015;16:100.
Stabley DL, Harris AW, Holbrook J, Chubbs NJ, Lozo KW, Crawford TO, et al. SMN1 and SMN2 copy numbers in cell lines derived from patients with spinal muscular atrophy as measured by array digital PCR. Mol Genet Genom Med. 2015;3:248–57.
Chien YH, Chiang SC, Weng WC, Lee NC, Lin CJ, Hsieh WS, et al. Presymptomatic diagnosis of spinal muscular atrophy through newborn screening. J Pediatr. 2017;190:124–9 e121.
Prior TW, Snyder PJ, Rink BD, Pearl DK, Pyatt RE, Mihal DC, et al. Newborn and carrier screening for spinal muscular atrophy. Am J Med Genet A. 2010;152A:1608–16.
Pyatt RE, Mihal DC, Prior TW. Assessment of liquid microbead arrays for the screening of newborns for spinal muscular atrophy. Clin Chem. 2007;53:1879–85.
Maranda B, Fan L, Soucy JF, Simard L, Mitchell GA. Spinal muscular atrophy: clinical validation of a single-tube multiplex real time PCR assay for determination of SMN1 and SMN2 copy numbers. Clin Biochem. 2012;45:88–91.
Gomez-Curet I, Robinson KG, Funanage VL, Crawford TO, Scavina M, Wang W. Robust quantification of the SMN gene copy number by real-time TaqMan PCR. Neurogenetics. 2007;8:271–8.
Zhang X, Wang B, Zhang L, You G, Palais RA, Zhou L, et al. Accurate diagnosis of spinal muscular atrophy and 22q11.2 deletion syndrome using limited deoxynucleotide triphosphates and high-resolution melting. BMC Genom. 2018;19:485.
Zhou L, Palais RA, Paxton CN, Geiersbach KB, Wittwer CT. Copy number assessment by competitive PCR with limiting deoxynucleotide triphosphates and high-resolution melting. Clin Chem. 2015;61:724–33.
Wang KC, Chang CC, Chang YF, Wang SH, Chiang CK, Tsai CP. Evaluation and characterization of a high-resolution melting analysis kit for rapid carrier-screening test of spinal muscular atrophy. J Neurogenet. 2015;29:113–6.
Cusco I, Barcelo MJ, del Rio E, Martin Y, Hernandez-Chico C, Bussaglia E, et al. Characterisation of SMN hybrid genes in Spanish SMA patients: de novo, homozygous and compound heterozygous cases. Hum Genet. 2001;108:222–9.
MacDonald WK, Hamilton D, Kuhle S. SMA carrier testing: a meta-analysis of differences in test performance by ethnic group. Prenat Diagn. 2014;34:1219–26.
Acknowledgements
We would like to thank the patients and their families for their contributions in this study. This work was supported by the Foundation for Young Scholars of Fujian Provincial Department of Health Office (project no. 2017-2-107), National Natural Science Foundation of China (project No. 81572084), and Xiamen Science and Technology Major Project (project No. 3502Z20171006).
Funding
This work was supported by the Foundation for Young Scholars of Fujian Provincial Department of Health Office (project no. 2017-2-107), National Natural Science Foundation of China (project No. 81572084), and Xiamen Science and Technology Major Project (project No. 3502Z20171006).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Xia, Z., Zhou, Y., Fu, D. et al. Carrier screening for spinal muscular atrophy with a simple test based on melting analysis. J Hum Genet 64, 387–396 (2019). https://doi.org/10.1038/s10038-019-0576-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s10038-019-0576-6
