
Abstract
Congenital heart defects (CHDs), the most common congenital human birth anomalies, involves complex genetic factors. Wnt/β-catenin pathway is critical for cardiogenesis and proved to be associated with numerous congenital heart abnormities. AXIN2 has a unique role in Wnt/β-catenin pathway, as it is not only an important inhibitor but also a direct target of Wnt/β-catenin pathway. However, whether AXIN2 is associated with human CHDs has not been reported. In our present study, we found a differential expression of Axin2 mRNA during the development of mouse heart, indicating its importance in mouse cardiac development. Then using targeted next-generation sequencing, we found two novel case-specific rare mutations [c.28 C > T (p.L10F), c.395 A > G (p.K132R)] in the sequencing region of AXIN2. In vitro functional analysis suggested that L10F might be a loss-of-function mutation and K132R is a gain-of-function mutation. Both mutations disrupted Wnt/β-catenin pathway and failed to rescue CHD phenotype caused by Axin2 knockdown in zebrafish model. Collectively, our study indicates that rare mutations in AXIN2 might contribute to the risk of human CHDs and a balanced canonical Wnt pathway is critical for cardiac development process. To our knowledge, it is the first study of AXIN2 mutations associated with human CHDs, providing new insights into CHD etiology.
Similar content being viewed by others


Introduction
Congenital heart defects (CHDs), the most prevalent congenital human birth anomalies with approximately 1∼5% frequency of newborns, are major public health issues in the world [1,2,3]. Although the etiology of CHD is complex, advances in human genetics and model animals development have provided us with a better understanding on genetic factors involving CHDs [4, 5]. Existing evidences have indicated an association between Wnt/β-catenin pathway and numerous congenital heart abnormities, such as aberrant atrioventricular canal development, tricuspid atresia, or defective outflow tract [6,7,8]. Activation of Wnt/β-catenin is required during gastrulation for induction of cardiac mesoderm, whereas its inhibition later enables cardiac terminal differentiation [9, 10]. Further researches have demonstrated that Wnt/β-catenin signaling is an important regulator of atrioventricular canal maturation at the initiation of valve development [6, 11].
Among the regulators of Wnt/β-catenin pathway, AXIN2 has a unique role. It is not only an important inhibitor of this pathway but also a direct target of Wnt activation, thus participating in a negative feedback loop to limit the duration or intensity of Wnt/β-catenin signaling. [12]. Such negative feedback loop allows AXIN2 to control this signaling precisely during development. Deletion of Axin2 in mice resulted in delayed heart valve maturation, with sustained active Wnt/β-catenin signaling [13]. A mutation of AXIN2 (V26D), which increased AXIN2 protein stability, was identified to cause mice heart abnormalities [14]. In addition, we found that Axin2 expressed higher during embryonic mice heart development while lower in adult heart tissues.
Based on above studies in mice, we predicate that AXIN2 is implicated in pathogenesis of CHD. To test this hypothesis, we sequenced all exons of AXIN2 in a CHD cohort with 417 cases and 213 matched controls. Two novel case-specific rare mutations [c.28 C > T (p.L10F), c.395 A > G (p.K132R)] were found in the coding region of AXIN2. In vitro and in vivo analyses showed that the two mutations have distinct roles in Wnt/β-Catenin pathway and CHD pathological process. This is the first report that mutations in AXIN2 are as likely to be associated with CHDs.
Methods
Ethics statement
All procedures performed in studies involving animals were in accordance with the ethical standards of the Institutional Animal Care and Use Committee of the School of Life Sciences, Fudan University.
Study subjects
Blood samples from 417 CHD patients (mean age 2.9 ± 2.7 years, 55.4% male) were collected between 2010 and 2012 from the Cardiovascular Disease Institute of Jinan Military Command (Jinan, China). CHD patients were diagnosed on the basis of echocardiography, with some further confirmed surgically. Detailed diagnosis information on the patients is shown in Table 1. All of the CHD cases were classified according to the method described previously [15]. The 213 controls (mean age 7.1 ± 3.7 years, 49.8% male) were ethnically gender-matched and unrelated volunteers without any congenital diseases recruited from the same geographical area. Patients with a positive family history of CHD in a first-degree relative or additional syndromes were excluded.
The present study was conducted in accordance with the Declaration of Helsinki principles. Protocols used in this work were reviewed and approved by the Ethics Committee of the School of Life Sciences, Fudan University, before the commencement of the study. Written informed consent was obtained from the parents and/or guardians of the children patients in this study.
DNA sequencing and data analysis
Approximately 2 ml of peripheral blood was collected from each test subject. Genomic DNA was prepared and targeted exome sequencing was conducted as previously described [16]. To confirm the genotyping results from next-generation sequencing, two case-specific coding mutations of AXIN2 were amplified by PCR and verified by Sanger DNA sequencing. The primers used in the PCR are listed in Supplementary Table 1.
All variants were checked in absence in ExAC database [17]. Missense mutations were also evaluated using PolyPhen2 [18], SIFT [19] MutationTaster [20] and MutationAssessor [21] for potentially damaging mutations.
Plasmid constructs and site-directed mutagenesis
AXIN2 plasmid was synthesized and inserted into the plasmid of pcDNA3.1(−)Myc-HisC by Generay Biotech (Shanghai, China). All mutations in AXIN2 were generated using a KOD Site-Directed Mutagenesis strategy (TOYOBO, Osaka, Japan). All plasmids used in this study were confirmed by DNA sequencing. The Topflash-luciferase reporter plasmid was a gift from Dr Tao Zhong at Fudan University.
Cell culture and treatment
HEK293T and SW480 cells were cultured using standard methods (ATCC). Wnt activation was achieved by treating cells with control or Wnt3a condition medium for 8 h before the collection of cells. Wnt3a condition medium was collected from Wnt3a secreting L cells as per the manufacturer’s instructions (ATCC).
Western blotting analysis
Myc-His-tagged wild-type or mutant AXIN2 was transfected into HEK293T cells. Thirty-six hours later, cells were lysed with RIPA buffer (1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS in 1 × PBS [pH 7.4]) containing a cocktail of protease inhibitors (Roche, Basel, Switzerland) and heated for 10 min at 100 °C. Cell lysates were loaded and separated on a 10% SDS-polyacrylamide gel electrophoresis and transferred onto a polyvinylidenedifluoride membrane (Merck Millipore, Darmstadt, Germany). After blocking for 1 h with 5% non-fat milk, the membrane was incubated with mouse Myc-Tag antibody (#2276, Cell Signaling Technology, Danvers, MA, USA) and mouse anti-β-actin antibody (A1978, Sigma-Aldrich, St. Louis, MO, USA) at 4 °C overnight. Horseradish peroxidase-conjugated anti-mouse IgG was served as secondary antibody (#7076, Cell signaling Technology, USA) for 2 h at room temperature and visualized with the ECL Detection System (Tanon, Shanghai, China). Band density was quantified with Image J and ratios of Myc/His AXIN2 relative to β-actin were used to measure the expression of wild-type and mutant AXIN2 protein. Three independent experiments were performed and representative results were shown.
Dual-luciferase reporter assay
HEK 293T cells were seeded (1.2 × 105) in 24-well cell culture plates 24 h before transfections and were transfected with 250 ng of wild-type/AXIN2 mutants/empty vector plasmids, 250 ng of Firefly-luciferase reporter (Topflash), and 10 ng of Renilla luciferase plasmid (pRLCMV) as an internal control, using Lipofectamine 2000 (Life Technologies, Waltham, MA, USA). Thirty-six hours post transfection, cells were treated with control or Wnt3a condition medium for 8 h. Cells were then lysed in Passive Lysis Buffer (Promega, Madison, WI, USA) and luciferase activity was analyzed using the dual-luciferase reporter assay system (Promega, USA) according to the manufacturer’s instructions. Three independent transfection experiments were performed and each luciferase assay was performed in triplicate.
Quantitative real-time PCR and reverse transcription PCR
Thirty-six hours post transfection, HEK293T cells were treated with control or Wnt3a condition medium for 8 h. Total RNA of cells and heart tissues from embryonic and adult mice was extracted by TRIzol and first-strand cDNA was synthesized. Quantitative real-time PCR (QRT-PCR) using SYBR Green was carried out to quantify mRNA changes of candidate Wnt/β-catenin/TCF target genes, including cMYC, LEF-1, and DKK1. Expression levels were calculated using the ΔΔCT method. Reverse transcription PCR (RT-PCR) was used to observe the expression pattern of Axin2 during the development of mouse heart. The primers for qRT-PCR and RT-PCR are listed in Supplementary Table 2.
Immunofluorescence and confocal microscopy
AXIN2 wild type or mutant was transfected into SW480 cells. Thirty-six hours post transfection, cells were treated with control or Wnt3a condition medium for 8 h. Cells were then fixed with 4% paraformaldehyde for 15 min and rendered permeable with 0.3% Triton X-100 for 5 min. After cells were blocked with 5% bovine serum albumin for 1 h at room temperature, they were incubated with rabbit anti-β-catenin antibody (ab32572, Abcam, Cambridge, MA, USA) and mouse Myc-Tag antibody (#2276, Cell Signaling Technology, USA) overnight at 4 °C in a humidified chamber. Then cells were washed with Tris-buffered saline and incubated with Cy3- or FITC-conjugated secondary antibodies (Life Technologies, USA) for 1 h at room temperature. All immunofluorescence (IF) micrographs were acquired using an Axiovert 200 M microscopy system (Carl Zeiss, Jena, Germany). Image quantifications were performed using Image J software.
Zebrafish injection
Wild-type AB strain zebrafish (Danio rerio) were maintained under standard conditions. Antisense Morpholino oligos (MOs) were designed to block the translation of Axin2 in zebrafish and were synthesized by Gene Tools. The sequences of Axin2 MOs and control MOs are as follows: Axin2 MO: 5′-GTTAGTGTCCTATTCATGGCTCTTG-3′ and control MO: 5′-CCTCTTACCTCAGTTACAATTTATA-3′. Three nanograms of Axin2 or control MO at volume of 2.3 nl were injected into each zebrafish embryo at one- to two-cell stage. In the rescue study, wild-type and mutant plasmids were linearized with Dra III-HF (New England Biolabs, Inc., Beverly, MA, USA) and transcribed with mMESSAGE mMACHINETM T7 (AM1344, Invitrogen, USA). The mRNAs were diluted in RNA-free water. Then, 200 pg human AXIN2-WT or AXIN2-MUT were co-injected with 3 ng Axin2 MO into each zebrafish embryo at one- or two-cell stage. Forty-eight hours post injection, photographs were taken of the collected embryos using a Zeiss 79005 Zen microscope and the percentage of abnormal embryos were calculated. Statistical significance was calculated using χ2- analysis.
Statistical analysis
Statistical analysis was performed using SPSS23.0. For in vitro analysis, data were presented as mean ± SD. Comparisons were performed by one-way analysis of variance among groups or by Student’s t-test between two groups. For in vivo data, statistical analysis was conducted using χ2-analysis. P < 0.05 were considered statistically significant.
Results
Identification of two novel AXIN2 mutations in Chinese CHD cases
To assess the role of AXIN2 in heart development, we measured the transcript level of Axin2 in mouse embryonic and adult heart. RT-PCR results revealed that Axin2 continues to express in mouse heart from embryonic to adulthood and has a specific developmental stage expression. Axin2 expression in mouse heart increases from embryonic day (E) 10.5–E12.5, reaching its maximum expression at E12.5, and then decreases from E15.5 to adulthood (Supplementary Figure 1). This result indicates its important role in mouse cardiac development.
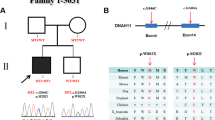
To investigate the potential role of AXIN2 in pathogenesis of CHDs, we sequenced all exons of AXIN2 in a CHD cohort with 417 cases and 213 matched controls. The detailed information of our CHD cohort is provided in Table 1. Two case-specific heterozygous missense mutations in AXIN2 [c.28 C > T (p.L10F), c.395 A > G (p.K132R)] were identified in two patients by targeted exome sequencing (Table 2) and further confirmed by Sanger sequencing (Fig. 1b). Both mutations were not identified in our 213 ethnically matched controls and were not reported in either ExAC database or any published papers. Cases with those two mutations were observed to have different CHD phenotypes (Table 2). Patient with mutation p.L10F was found to have trilogy of fallot (TOF) and the other case with mutation p.K132R displayed mitral insufficiency (Supplementary Figure 2). Meanwhile, no missense mutations in other common genes associated with CHD such as NKX2.5/CSX, GATA4, TBX1, and TBX5 were identified in both cases. L10F is located near the tankyrase-binding region and K132R is located in the regulator of G protein signaling (RGS) domain, which is a highly conserved region (Fig. 1a). Both mutations occurred in the highly conserved amino acids (Fig. 1c) and are predicted to be damaging by PolyPhen2 and MutationTaster (Table 2).

Rare mutations of AXIN2 identified in human CHD cases. a A schematic diagram of the AXIN2 protein structure and the location of the missense mutations. The predicted interaction domains of AXIN2 protein are depicted with colors. APC adenomatous polyposis coli, DIX dimerization motif, GSK3β glycogen synthase kinase 3β, TNKS tankyrase. b Sanger sequencing results showed heterozygous missense mutations c.28 C > T (p.L10F) and c.395 A > G (p.K132R). c A partial alignment of human AXIN2 with eight other orthologous sequences. Both AXIN2 mutations found in CHD patients occurred in the highly conserved amino acids (indicated by arrows)
The K132R mutant upregulated AXIN2 protein expression, whereas L10F mutant had no significant effects on protein expression
To investigate whether these missense mutations of AXIN2 resulted in protein expression change, we transfected the two mutant AXIN2 plasmids as well as wild-type AXIN2 plasmids into HEK293T cells, respectively, and detect protein expression by use of western blot analysis. The results demonstrated that compared with wild-type, L10F mutant had no significant effects on the protein expression of AXIN2, whereas K132R significantly increased AXIN2 protein expression, making its expression almost doubled (Fig. 2).

Western blot analysis of AXIN2 protein. HEK293T cells were transfected with Myc-His-tagged wild-type or mutant AXIN2. Thirty-six hours later, cell lysates were blotted with a Myc-Tag antibody. The representative images were shown in a and the statistical analysis were shown in b. Three independent experiments were performed and the data are presented as mean ± SD (*p < 0.05 vs. WT)
L10F and K132R mutants affected Wnt/β-catenin signaling
One of the primary functions of AXIN2 is to promote the turnover of β-catenin, thus inhibiting the Wnt/β-catenin signaling as well as the expression of downstream target genes [22]; therefore, we used luciferase reporter assay to analyze whether these mutations affected the activation level of Wnt/β-catenin pathway in HEK293T cells. As expected, Wnt3a treatment in HEK293T cells increased the activity of the β-catenin-responsive reporter, TOPFlash, by almost fivefold, whereas ectopic overexpression of both wild-type and two mutant AXIN2 proteins inhibited TOPFlash reporter gene activity. However, compared with wild-type AXIN2, L10F mutant significantly upregulated Wnt/β-catenin signaling, whereas K132R mutant resulted in a significantly lower activation of Wnt/β-catenin (Fig. 3a).

Effects of AXIN2 variants on Wnt/β-catenin pathway. a Analysis of relative luciferase reporter activity. HEK293T cells were transfected with empty vector (as control), WT, or mutant AXIN2 expression constructs, as well as TOPFlash reporter vector. Renilla luciferase plasmid was cotransfected as an internal control. Thirty-six hours after transfection, cells were treated with control or Wnt3a condition medium for 8 h before being collected. Relative luciferase activity was reported as the ratio of TopFlash/Renilla luciferase activity. b Cells were collected and total RNA was extracted. Expressions of the candidate Wnt/β-catenin/TCF target genes were assessed by qRT-PCR. c Representative images of immunofluorescence staining for β-catenin (green) and Myc-tagged AXIN2 (red) in SW480 cells. The nuclei were stained with 4’, 6-diamidino-2-phenylindole (DAPI) (blue). d Immunofluorescence intensity of β-catenin in SW480 cells, which were transfected with AXIN2. All experiments were performed in triplicate and the data are presented as mean ± SD. (&&p < 0.01 vs. Vector, *p < 0.05 vs. Vector + Wnt3a, **p < 0.01 vs. Vector + Wnt3a, #p < 0.05 vs. WT + Wnt3a, ##p < 0.01 vs. WT + Wnt3a)
We then assessed the effects of wild-type and mutant AXIN2 on the expression of several candidate Wnt/β-catenin/TCF target genes, including cMYC, LEF-1, and DKK1. QRT-PCR results revealed that all those target genes were induced by Wnt3a treatment and were significantly inhibited by ectopic AXIN2 overexpression. Besides, compared with wild-type AXIN2, L10F mutant upregulated the expression of the target genes, whereas K132R mutant AXIN2 significantly reduced them (Fig. 3b). Therefore, the qRT-PCR results were consistent with the luciferase reporter assay results. These results indicated that L10F might be a loss-of-function mutation, whereas K132R is a gain-of-function mutation.
L10F and K132R mutations affected the ability of AXIN2 to degrade β-catenin
AXIN2 is an important component of the destruction complex, which degrades β-catenin; therefore, changes of β-catenin expression caused by mutations of AXIN2 were observed. IF was also carried out with SW480 cells, in which β-catenin is extraordinarily stable due to adenomatous polyposis coli (APC) mutation. As expected, results showed that AXIN2 induced the degradation of β-catenin in SW480 cells. Compared with wild-type AXIN2, L10F mutant resulted in the accumulation of β-catenin in the cytoplasm. As for the K132R mutant, it resulted in a further degradation of intracellular β-catenin compared with wild-type AXIN2 (Fig. 3c, D).
L10F and K132R mutations failed to rescue Axin2-mediated CHD phenotype in zebrafish
Next, we explored the potential in vivo pathogenetic effect of L10F and K132R mutations in zebrafish. About 73% of zebrafish injected with antisense MOs for zebrafish Axin2 displayed an enlarged pericardium (a CHD phenotype) (Fig. 4a) and this Axin2-mediated CHD phenotype could be partially rescued by co-injected with human wild-type AXIN2 mRNA (Fig. 4b). However, both L10F and K132R mutations failed to rescue Axin2-mediated CHD phenotype in zebrafish (Fig. 4b).

AXIN2 mutations failed to rescue Axin2-mediated CHD phenotype in zebrafish. a Pericardium defects were observed in zebrafish embryos after Axin2 MO (3 ng) injection. Red arrows indicate the pericardial cavity of zebrafish embryos. b The response frequencies of pericardial abnormalities. Co-injection of 200 pg wild-type AXIN2 mRNA partially rescued Axin2 MO-mediated CHD phenotype (enlarged pericardium), whereas co-injected 200 pg AXIN2 L10F and K132R mRNA had no effect at all. The number above each bar is the total number of embryos examined under each experimental condition. P-value was calculated using χ2-analysis. (**p < 0.01, ns not significant)
Discussion
In the present study, we identified two novel case-specific rare mutations (L10F, K132R) of AXIN2 in Chinese CHD cases. Our results demonstrated that both mutations affected in vitro and in vivo functions of AXIN2, resulting in significant changes of Wnt/β-catenin signaling and inducing cardiac defects. Therefore, our data provide evidence that rare mutations in AXIN2 might contribute to the risk of human CHDs. To our knowledge, this is the first report of AXIN2 mutations associated with human CHDs.
AXIN2, a key negative regulator of Wnt/β-catenin signaling, has been suggested to have an important role during embryogenesis [23, 24]. In the present study, we used a Wnt3a-responsive luciferase reporter assay and demonstrated that activation of the Wnt/β-catenin signaling by Wnt3a could be inhibited by overexpression of wild-type AXIN2, indicating that AXIN2 is capable of antagonizing canonical Wnt signaling, which is consistent with previous studies [25].
Mutations in AXIN2 have been found to be associated with human diseases such as oligodontia and colorectal cancer [22, 26], however, whether it is responsible for human CHD has not been reported before. Previous studies have reported the relationship between mutations of other Wnt/β-catenin-related genes and human CHDs [27, 28]; therefore, we proposed that AXIN2 may also have a role in CHD development.
In the present study, we discovered two novel case-specific mutations of AXIN2 in human CHD for the first time and both mutations are likely to affect the function of AXIN2. The first mutation L10F might be a loss-of-function mutation. Although it has no effect on AXIN2 protein expression, this mutation caused significant upregulation of Wnt/β-catenin signaling compared with wild-type AXIN2, leading to elevated target genes and the accumulation of β-catenin. Therefore, the inhibition ability of Wnt/β-catenin signaling might be blocked because of L10F mutation of AXIN2. The other mutant K132R caused a lysine-to-arginine substitution, which is located in the conserved RGS domain and resulted in further inhibition of Wnt/β-catenin signaling and the expression of Wnt target genes. Therefore, it might be a gain-of-function mutation. As K132R significantly upregulated the protein expression of AXIN2, we speculate that the inhibition of Wnt/β-catenin signaling might be partly due to AXIN2 protein change. The increase in AXIN2 protein could be caused by increased protein stability or increased mRNA level of AXIN2, which is to be investigated in our further research. On the other hand, as K132R is located on the APC-binding domain (Fig. 1a), it is also likely that the mutation enhanced the interaction between AXIN2 and APC, thus inhibiting the downstream Wnt/β-catenin signaling. The detailed mechanism regarding how K132R affected the Wnt/β-catenin signaling remains further elucidated. Despite opposite effects of the two mutations on Wnt/β-catenin signaling, in vivo evidence demonstrated that both L10F and K132R failed to rescue the CHD phenotype caused by Axin2 knockdown in zebrafish. Therefore, a balanced canonical Wnt pathway is of crucial importance during cardiac development process.
In conclusion, we found two rare mutations of AXIN2 (L10F and K132R) in CHD cases, which significantly affected Wnt/β-catenin signaling. For the first time, our findings indicate that genetic variation in AXIN2 gene may be associated with CHD risk. Our results also strongly suggested that a balanced canonical Wnt pathway is critical for cardiac development process, as not only loss-of- or gain-of-function mutations of AXIN2 might disturb Wnt/β-catenin signaling, thus inducing CHD.
References
Wessels MW, Willems PJ. Genetic factors in non-syndromic congenital heart malformations. Clin Genet. 2010;78:103–23.
Hoffman JI, Kaplan S. The incidence of congenital heart disease. J Am Coll Cardiol. 2002;39:1890–900.
van der Linde D, Konings EE, Slager MA, Witsenburg M, Helbing WA, Takkenberg JJ, et al. Birth prevalence of congenital heart disease worldwide: a systematic review and meta-analysis. J Am Coll Cardiol. 2011;58:2241–7.
Jenkins KJ, Correa A, Feinstein JA, Botto L, Britt AE, Daniels SR, et al. Noninherited risk factors and congenital cardiovascular defects: current knowledge: a scientific statement from the American Heart Association Council on Cardiovascular Disease in the Young: endorsed by the American Academy of Pediatrics. Circulation. 2007;115:2995–3014.
Pierpont ME, Basson CT, Benson DW Jr, Gelb BD, Giglia TM, Goldmuntz E, et al. Genetic basis for congenital heart defects: current knowledge: a scientific statement from the American Heart Association Congenital Cardiac Defects Committee, Council on Cardiovascular Disease in the Young: endorsed by the American Academy of Pediatrics. Circulation. 2007;115:3015–38.
Gillers BS, Chiplunkar A, Aly H, Valenta T, Basler K, Christoffels VM, et al. Canonical wnt signaling regulates atrioventricular junction programming and electrophysiological properties. Circ Res. 2015;116:398–406.
Cohen ED, Wang Z, Lepore JJ, Lu MM, Taketo MM, Epstein DJ, et al. Wnt/beta-catenin signaling promotes expansion of Isl-1-positive cardiac progenitor cells through regulation of FGF signaling. J Clin Invest. 2007;117:1794–804.
Jeong MH, Kim HJ, Pyun JH, Choi KS, Lee DI, Solhjoo S, et al. Cdon deficiency causes cardiac remodeling through hyperactivation of WNT/beta-catenin signaling. Proc Natl Acad Sci USA. 2017;114:E1345–e54.
Tian Y, Cohen ED, Morrisey EE. The importance of Wnt signaling in cardiovascular development. Pediatr Cardiol. 2010;31:342–8.
Gessert S, Kuhl M. The multiple phases and faces of wnt signaling during cardiac differentiation and development. Circ Res. 2010;107:186–99.
Bosada FM, Devasthali V, Jones KA, Stankunas K. Wnt/beta-catenin signaling enables developmental transitions during valvulogenesis. Development. 2016;143:1041–54.
Jho EH, Zhang T, Domon C, Joo CK, Freund JN, Costantini F. Wnt/beta-catenin/Tcf signaling induces the transcription of Axin2, a negative regulator of the signaling pathway. Mol Cell Biol. 2002;22:1172–83.
Hulin A, Moore V, James JM, Yutzey KE. Loss of Axin2 results in impaired heart valve maturation and subsequent myxomatous valve disease. Cardiovasc Res. 2017;113:40–51.
Qian L, Mahaffey JP, Alcorn HL, Anderson KV. Tissue-specific roles of Axin2 in the inhibition and activation of Wnt signaling in the mouse embryo. Proc Natl Acad Sci USA. 2011;108:8692–7.
Botto LD, Lin AE, Riehle-Colarusso T, Malik S, Correa A. Seeking causes: Classifying and evaluating congenital heart defects in etiologic studies. Birth Defects Res A Clin Mol Teratol. 2007;79:714–27.
Qiao X, Liu Y, Li P, Chen Z, Li H, Yang X, et al. Genetic analysis of rare coding mutations in CELSR1-3 in Chinese Congenital Heart and Neural Tube Defects. Clin Sci (Lond) 2016;2329–2340.
Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–91.
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–9.
Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–81.
Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014;11:361–2.
Reva B, Antipin Y, Sander C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res. 2011;39:e118.
Mazzoni SM, Petty EM, Stoffel EM, Fearon ER. An AXIN2 mutant allele associated with predisposition to colorectal neoplasia has context-dependent effects on AXIN2 protein function. Neoplasia. 2015;17:463–72.
Alldredge A, Fuhrmann S. Loss of Axin2 causes ocular defects during mouse eye development. Invest Ophthalmol Vis Sci. 2016;57:5253–62.
Eckei G, Boing M, Brand-Saberi B, Morosan-Puopolo G. Expression pattern of Axin2 during chicken development. PLoS ONE. 2016;11:e0163610.
Koch A, Hrychyk A, Hartmann W, Waha A, Mikeska T, Waha A, et al. Mutations of the Wnt antagonist AXIN2 (Conductin) result in TCF-dependent transcription in medulloblastomas. Int J Cancer. 2007;121:284–91.
Lammi L, Arte S, Somer M, Jarvinen H, Lahermo P, Thesleff I, et al. Mutations in AXIN2 cause familial tooth agenesis and predispose to colorectal cancer. Am J Hum Genet. 2004;74:1043–50.
Pu Y, Chen P, Zhou B, Wang Y, Song Y, Peng Y, et al. Association between polymorphisms in AXIN1 gene and atrial septal defect. Biomarkers. 2014;19:674–8.
Furtado MB, Wilmanns JC, Chandran A, Perera J, Hon O, Biben C, et al. Point mutations in murine Nkx2-5 phenocopy human congenital heart disease and induce pathogenic Wnt signaling. JCI Insight. 2017;2:e88271.
Acknowledgements
We appreciate the patient family for their collaboration in this study. We thank Professor Tao Zhong for his plasmid that was used in this study. This study was funded by grants from the National Natural Science Foundation of China (81670973, 31601029, and 81601298) and China Postdoctoral Science Foundation (BX20180069 and 2018M641918).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Zhu, MJ., Ma, XY., Ding, PC. et al. Novel mutations of AXIN2 identified in a Chinese Congenital Heart Disease Cohort. J Hum Genet 64, 427–435 (2019). https://doi.org/10.1038/s10038-019-0572-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s10038-019-0572-x
This article is cited by
-
DNA methylation and cardiovascular disease in humans: a systematic review and database of known CpG methylation sites
Clinical Epigenetics (2023)
-
Understanding Heart Field Progenitor Cells for Modeling Congenital Heart Diseases
Current Cardiology Reports (2021)
