
Abstract
The genetic causes of combined pituitary hormone deficiency remain elusive in most patients. Recently, incompletely penetrant heterozygous mutations in ROBO1 have been described in patients with pituitary stalk interruption syndrome. Herein, we identified a novel homozygous slice site mutation in ROBO1 (c.1342+1G>A) using a trio whole-exome sequencing strategy in a 5-year-old Japanese boy who had combined pituitary hormone deficiency, psychomotor developmental delay, severe intellectual disability, sensorineural hearing loss, strabismus, and characteristic facial features, including a broad forehead, micrognathia, and arched eyebrows. Magnetic resonance imaging delineated anterior pituitary hypoplasia, ectopic posterior pituitary, invisible pituitary stalk, thinning of the corpus callosum, and hypoplasia of the pons and midbrain. The phenotypically normal parents (first cousins) were heterozygous for the mutation. The results provide further evidence of ROBO1 being involved in the development of the pituitary gland. A recessive mutation of ROBO1 is a potential novel cause of a syndromic disorder associated with combined pituitary hormone deficiency.
Similar content being viewed by others

Introduction
Normal pituitary development requires a complex genetic cascade of transcription factors and signaling molecules, either intrinsic or extrinsic to the developing Rathke’s pouch [1]. Mutations of genes involved in these processes, including POU1F1, PROP1, HESX1, LHX3, LHX4, OTX2, GLI2, and SOX2, are associated with a wide range of pituitary phenotypes, such as isolated growth hormone (GH) deficiency and combined pituitary hormone deficiency (CPHD), which is defined as the presence of hormone deficits affecting at least two anterior pituitary hormone lineages [1, 2]. However, the definitive genetic causes remain obscure in the majority of patients with CPHD [2, 3].
Congenital hypopituitarism is frequently associated with other extrapituitary abnormalities, such as anophthalmia/microphthalmia, optic nerve hypoplasia, dysgenesis of the corpus callosum, absence of the septum pellucidum, and holoprosencephaly, suggesting that defects in signaling molecules or transcription factors involved in the development of the forebrain result in such syndromic disorders [4].
The roundabout guidance receptors (ROBOs) and their Slit guidance ligands play critical roles in axonal guidance, which is essential for the formation of the neuronal network in the central nervous system. ROBO1 acts as the gatekeeper controlling the midline crossing of axons [5].
In the present study, we identified a homozygous splice-acceptor site mutation in the ROBO1 gene in a patient with a characteristic syndromic disorder associated with CPHD. Our study implies that recessive ROBO1 null mutations cause a novel neurodevelopmental syndrome associated with CPHD.
Materials and methods
Case reports
This Japanese male patient was born at 38 weeks of gestation as the first child of consanguineous phenotypically normal parents (first cousins) with no other significant family history. At birth, his length was 50.0 cm (+0.9 standard deviation [SD]), his weight 3.28 kg (+1.2 SD), and his head circumference 37.5 cm (+3.4 SD). He had distinct facial features with a broad forehead, micrognathia, a broad philtrum, and arched eyebrows (Fig. 1a). He also had hypotonia, micropenis, cryptorchidism, strabismus, and sensorineural deafness. Brain magnetic resonance imaging delineated hydrocephalus, anterior pituitary hypoplasia, ectopic posterior pituitary, invisible pituitary stalk, thinning of the corpus callosum, and hypoplasia of the pontine and midbrain (Fig. 1b, c).

Clinical findings in the patient. a A front view of the patient at one year of age showing distinct facial features with strabismus, a broad forehead, micrognathia, a broad philtrum, and arched eyebrows. T1-weighted sagittal (b) and coronal (c) views of the brain magnetic resonance imaging show anterior pituitary hypoplasia, ectopic posterior pituitary (white arrow), thinning of the corpus callosum, and the pontine and the midbrain hypoplasia. d Diffusion tensor imaging and fiber tractography showing the presence of transverse pontine fibers. The authors have obtained informed consent from his parents to publication of these images
At 20 days of age, he developed recurrent hypoglycemia and conjugated hyperbilirubinemia. A hormonal examination for critical samples obtained at the time of spontaneous presentation of hypoglycemia showed central hypothyroidism and low serum cortisol and plasma ACTH levels, suggesting the presence of CPHD (Table 1). He was therefore started on thyroid hormone and hydrocortisone replacement therapies. At 18 months of age, his height was 64.5 cm (−5.1 SD). Endocrine studies at that time confirmed the diagnosis of CPHD (associated with deficiencies of GH, TSH, prolactin, LH, FSH, and ACTH) (Table 1), and recombinant human GH therapy was started.
At the final examination at 5 years of age, his motor and mental development was severely retarded. He was unable to speak any meaningful words and sit alone. Diffusion tensor imaging and fiber tractography, performed after the genetic diagnosis, demonstrated thinning of the corpus callosum and the anterior commissure but showed the presence of transverse pontine fibers (Fig. 1d).
Molecular studies
This study was approved by the Institutional Review Board at Nagasaki University Graduate School of Biomedical Sciences. Trio whole-exome sequencing was performed using a SureSelect Human All Exon V5 (Agilent Technologies, Santa Clara, CA, USA) on a HiSeq 2500 platform (Illumina, San Diego, CA, USA). DNA was obtained from peripheral blood samples of the patient and the parents after written informed consent was obtained from the parents. The reads in the FASTQ files were aligned to the human reference genome using Novoalign version 3.0 (http://www.novocraft.com/). The mean depth of the RefSeq coding region was 140.54% with 97.2% of total coding sequences covered by 20 reads or more in the proband. Trio-based genomic variation information was detected by the Genome Analysis Toolkit software version 3.4-46 [6]. Subsequently, de novo, homozygous, compound heterozygous, and X-linked variations in exons and canonical splice sites (±2 bp) were extracted and annotated by the ANNOVAR software [7]. This process excluded variants with allele frequencies > 0.5% in any of the Exome Aggregation Consortium (ExAC) (http://exac.broadinstitute.org/), NHLBI GO Exome Sequencing Project (http://evs.gs.washington.edu/EVS/), Human Genetic Variation Database (http://www.hgvd.genome.med.kyoto-u.ac.jp), the 1KJPN database of Tohoku Medical Megabank (http://www.dist.megabank.tohoku.ac.jp), and in-house exome data. Heterozygous variations sharing the same GENCODE v19 genes were also extracted to detect compound heterozygous mutations. The candidate variants identified in the strategy were confirmed via Sanger sequencing.
Results
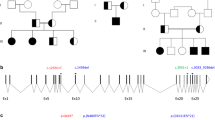
Using the trio-based strategy and the filtering methods, we identified eight candidate variants consisting of six homozygous and two X-linked hemizygous variants (Supplemental Table 1). Of these, a homozygous splice-acceptor site mutation in ROBO1 (c.1342+1G>A, NM_002941) was proposed as the best candidate by the Online Mendelian Inheritance in Man database information of known diseases (www.omim.org) (Fig. 2a, b). This splice mutation is predicted to cause exon skipping and frameshift mediating nonsense-mediated mRNA decay (http://www.fruitfly.org/seq_tools/splice.html) [8]. The father and the mother of the proband were heterozygous for the mutation. The patient had no other pathogenic mutations in genes known to cause CPHD [2].

Mutation analyses of ROBO1 in this family. a Electrochromatograms delineating the homozygous mutation in a splice-acceptor site (c.1342+1G>A, NM_002941) in the patient (asterisk) and the heterozygous ones in the parents. The mutation was confirmed by direct sequencing. b Pedigree of the family. The black-painted square indicates the presence of the homozygous variant. Half-black, half-white symbols represent carriers of the variant in a heterozygous form
Discussion
We identified a homozygous ROBO1 splice-acceptor site mutation in a patient with syndromic CPHD and summarized the genetic and clinical features of patients previously reported to have ROBO1 mutations (Table 2). To our knowledge, the combination of his clinical manifestations has not been reported thus far. Therefore, we propose the null homozygous mutation of ROBO1 as the likely genetic cause of a novel syndrome associated with CPHD, based on the following: First, Robo1 null mice, which die shortly after birth, show defects in axon pathfinding with dysgenesis of the corpus callosum and the hippocampal commissure [5], phenotypically similar to those of patients with biallelic ROBO1 mutations (the present patient and Case 6 in Table 2). Second, heterozygous mutations in ROBO1 have been recently reported in five patients (Cases 1 through 5 in Table 2) from three independent families with pituitary stalk interruption syndrome [9] and variable pituitary phenotypes ranging from isolated GH deficiency to CPHD, indicating that ROBO1 is involved in the pituitary development and function (Table 2). However, the penetrance of the dominant ROBO1 mutations seems to be incomplete, as phenotypically normal members in the pedigrees also had the same mutation. Indeed, the present parents, harboring a heterozygous ROBO1 mutation, seem phenotypically normal. Furthermore, heterozygous ROBO1 loss-of-function variants, including nonsense, frameshift, and splice site mutations, are described in the ExAC database. Third, not only homozygous but also heterozygous patients exhibit various ophthalmological phenotypes, such as strabismus, optic nerve hypoplasia, and hypermetropia (Table 2) [9, 10]. This may not be surprising, considering that Robo/Slit signaling plays a critical role in the extension of the retinal ganglion cell axons from the eye to the brain and formation of the optic chiasm [11]. Fourth, a patient with biallelic compound heterozygous missense variants in ROBO shared some phenotypes with the present patient, such as intellectual disability and thinning of the anterior commissure and corpus callosum [10]. However, the previously reported patient did not exhibit any abnormalities of the pituitary gland, indicating that the pituitary phenotypes in patients with biallelic ROBO1 mutations may be variable. Taken together, these findings imply that the homozygous ROBO1 null mutations cause a characteristic neurodevelopmental disorder with CPHD and defects in axon pathfinding, and heterozygous mutations may also cause diverse clinical features, ranging from nearly normal to pituitary stalk interruption syndrome and showing a wide range of penetrance, with expressivity depending on other genetic and environmental factors.
The pathological mechanisms of invisible stalk and pituitary dysfunction in patients with ROBO1 mutations remain obscure. Since ROBO1 defects possibly lead to abnormal axon elongation of magnocellular neurons from the paraventricular and supraoptic nuclei of the hypothalamus to the posterior pituitary, ROBO1 mutations may affect close relationship and tissue interactions between oral and neural ectoderm, which are critical for development and differentiation of the pituitary gland. Therefore, it is reasonable to hypothesize that ROBO1 mutations result in pituitary dysmorphogenesis and dysfunction [1].
In conclusion, the results provide further evidence of the involvement of ROBO1 in the pituitary development. Recessive null mutations of ROBO1 may cause novel syndromic CPHD. At this time, however, the phenotypic spectrum and mechanisms underlying the development of pituitary dysfunction remain to be determined in patients with ROBO1 mutations. These matters await further investigations.
References
Kelberman D, Rizzoti K, Lovell-Badge R, Robinson IC, Dattani MT. Genetic regulation of pituitary gland development in human and mouse. Endocr Rev. 2009;30:790–829.
Fang Q, George AS, Brinkmeier ML, Mortensen AH, Gergics P, Cheung LY, et al. Genetics of combined pituitary hormone deficiency: roadmap into the genome era. Endocr Rev. 2016;37:636–75.
Dateki S, Fukami M, Uematsu A, Kaji M, Iso M, Ono M, et al. Mutation and gene copy number analyses of six pituitary transcription factor genes in 71 patients with combined pituitary hormone deficiency: identification of a single patient with LHX4 deletion. J Clin Endocrinol Metab. 2010;95:4043–7.
Kelberman D, Dattani MT. Role of transcription factors in midline central nervous system and pituitary defects. Endocr Dev. 2009;14:67–82.
Andrews W, Liapi A, Plachez C, Camurri L, Zhang J, Mori S, et al. Robo1 regulates the development of major axon tracts and interneuron migration in the forebrain. Development. 2006;133:2243–52.
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–303.
Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164.
Holbrook JA, Neu-Yilik G, Hentze MW, Kulozik AE. Nonsense-mediated decay approaches the clinic. Nat Genet. 2004;36:801–8.
Bashamboo A, Bignon-Topalovic J, Moussi N, McElreavey K, Brauner R. Mutations in the human ROBO1 gene in pituitary stalk interruption syndrome. J Clin Endocrinol Metab. 2017;102:2401–6.
Calloni SF, Cohen JS, Meoded A, Juusola J, Triulzi FM, Huisman TAGM, et al. Compound heterozygous variants in ROBO1 cause a neurodevelopmental disorder with absence of transverse pontine fibers and thinning of the anterior commissure and corpus callosum. Pediatr Neurol. 2017;70:70–4.
Thompson H, Barker D, Camand O, Erskine L. Slits contribute to the guidance of retinal ganglion cell axons in the mammalian optic tract. Dev Biol. 2006;296:476–84.
Acknowledgements
We thank the patient and his parents for participating in this study. We also thank Tadashi Matsumoto and Tatsuro Kondo from Misakaenosono Mutsumi Developmental, Medical, and Welfare Center; and Akiko Nakatomi, Manami Ishibashi, Akinori Yanai, Tsutomu Ogata, Toshiharu Kihara, and Keishi Tsunoda from Nagasaki University Hospital for their help in collecting clinical information and for participating in fruitful discussions. This work was supported by a grant for the Initiative on Rare and Undiagnosed Diseases in Pediatrics (no. 18gk0110012h0101) from the Japan Agency for Medical Research and Development (AMED), Tokyo, Japan, and by Grants-in-Aid for Young Scientists (B) (25860871) from the Ministry of Education, Culture, Sports, Science, and Technology, Japan.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Dateki, S., Watanabe, S., Mishima, H. et al. A homozygous splice site ROBO1 mutation in a patient with a novel syndrome with combined pituitary hormone deficiency. J Hum Genet 64, 341–346 (2019). https://doi.org/10.1038/s10038-019-0566-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s10038-019-0566-8
