
Abstract
Casein kinase 2 (CK2) is a serine threonine kinase ubiquitously expressed in eukaryotic cells and involved in various cellular processes. In recent studies, de novo variants in CSNK2A1 and CSNK2B, which encode the subunits of CK2, have been identified in individuals with intellectual disability syndrome. In this study, we describe four patients with neurodevelopmental disorders possessing de novo variants in CSNK2A1 or CSNK2B. Using whole-exome sequencing, we detected two de novo variants in CSNK2A1 in two unrelated Japanese patients, a novel variant c.571C>T, p.(Arg191*) and a recurrent variant c.593A>G, p.(Lys198Arg), and two novel de novo variants in CSNK2B in Japanese and Malaysian patients, c.494A>G, p.(His165Arg) and c.533_534insGT, p.(Pro179Tyrfs*49), respectively. All four patients showed mild to profound intellectual disabilities, developmental delays, and various types of seizures. This and previous studies have found a total of 20 CSNK2A1 variants in 28 individuals with syndromic intellectual disability. The hotspot variant c.593A>G, p.(Lys198Arg) was found in eight of 28 patients. Meanwhile, only five CSNK2B variants were identified in five individuals with neurodevelopmental disorders. We reviewed the previous literature to verify the phenotypic spectrum of CSNK2A1- and CSNK2B-related syndromes.
Similar content being viewed by others


Introduction
Neurodevelopmental disorders (NDDs) are a group of neuropsychiatric deficits diagnosed during early childhood. They include intellectual disability, autism spectrum disorder, attention deficit/hyperactivity disorder, specific learning disorders, communication disorders, motor disorders, and tic disorders [1]. It is estimated that approximately 2–5% of children may be affected by NDDs [2,3,4]. Recent trio-based whole-exome sequencing (WES) analyses have revealed that de novo variants in loss-of-function intolerant or haploinsufficient genes are important causative factors of NDDs [5,6,7,8,9]. However, the genetic causes of NDDs are heterogeneous and have not been fully elucidated.
Protein kinase CK2 is a pleiotropic serine/threonine kinase phosphorylating hundreds of substrates and participating in diverse cellular processes, including cell cycle regulation [10, 11], DNA replication and repair [12], development and differentiation [13], transcription [14], cell signaling [15], carcinogenesis [16], and apoptosis [17]. CK2 acts as a tetrameric complex comprising two catalytic (CK2α and/or CK2α′) and two non-catalytic (CK2β) subunits, and CK2 holoenzyme comprises identical (α/α or α′/α′) or non-identical (α/α′) combinations of catalytic subunits [18,19,20,21,22]. Recent WES studies have reported that de novo variants in CSNK2A1 and CSNK2B, which encode CK2α and CK2β subunits, respectively, cause NDDs [23,24,25,26,27,28,29]. The disorder caused by CSNK2A1 variants is known as Okur-Chung neurodevelopmental syndrome (MIM#617062), and is characterized by intellectual disabilities, developmental delays, behavioral problems, and other multisystemic abnormalities [23,24,25,26, 29]. To date, 26 individuals with de novo variants in CSNK2A1 have been reported. In contrast, only three individuals harboring de novo variants in CSNK2B have been identified with intellectual disabilities, developmental delays, and myoclonic epilepsy [27, 28].
Here, we describe four individuals exhibiting intractable epilepsy, mild to severe psychomotor retardation, and intellectual disability. Using WES, we identified two de novo CSNK2A1 variants and two CSNK2B variants in these patients. We will describe detailed clinical features of patients with CSNK2A1 and CSNK2B variants to delineate the phenotypic spectrum of CK2-related syndromes.
Materials and methods
Patients
A total of 1230 individuals with early childhood-onset epilepsy and their parents were investigated in this study. Written informed consent was obtained from all participating families and the genomic DNA were extracted from blood leukocytes. Detailed clinical information was obtained from corresponding clinicians. Experimental protocols were approved by the Institutional Review Board of Yokohama City University School of Medicine, Showa University Faculty of Medicine, and Hamamatsu University School of Medicine.
Whole-exome sequencing (WES)
Trio-based WES of 337 families and case-only WES of 893 individuals was performed using the SureSelectXT Human All Exon v4, v5 or v6 (Agilent Technologies, Santa Clara, CA). Captured libraries were sequenced using Illumina HiSeq 2000 or 2500 (Illumina, San Diego, CA) with 101-base paired-end reads. Exome data processing, variant calling, and variant annotation were performed as previously described [30,31,32]. Final variants were annotated with Annovar [33] for predictive value of functional impact of the coding variants and assessing allele frequency: in-house database of 575 control exomes, 1KJPN [34] 1000 genome [35] and ExAC database [36]. Variant pathogenicity was predicted by SIFT, Polyphen-2, CADD [37] and M-CAP [38] software. Nucleotide conservation prediction was performed using GERP and PhastCons. Candidate variants were confirmed by Sanger sequencing using ABI 3130xl Genetic Analyzer (Applied Biosystems, Foster City, CA).
Plasmid construction
Human WT CSNK2A1 (NM_177559.2) and CSNK2B (NM_001320.5) cDNAs were prepared by polymerase chain reaction using a cDNA library transcribed from mRNA derived from human brain tissues as a template, and verified by Sanger sequencing. WT CSNK2B cDNA was cloned into a pCAGGS-IRES2-nucEGFP (pCIG) vector to express N-terminal HA-tagged CSNK2B, as well as nuclear-localized EGFP. Site-directed mutagenesis using a KOD-Plus-Mutagenesis kit (TOYOBO, Osaka, Japan) was used to generate mutant CSNK2B (p.Thr37Tyrfs*5, p.His165Arg, and p.Pro179Tyrfs*49) vectors in accordance with the manufacturer’s instructions. WT CSNK2A1 cDNA was cloned into a pEF1α-1xFLAG vector to express N-terminal FLAG-tagged CSNK2A1 under an EF1α promoter.
Cell culture, transfection, immunoprecipitation, and western blotting
HEK293T human embryonic kidney cells were grown in DMEM supplemented with 4.5 g/l glucose, l-glutamine (Wako, Osaka, Japan, 043-30085), 10% FBS, and penicillin/streptomycin at 37 °C and 5% CO2.
WT or mutant HA-tagged CSNK2B vectors with/without FLAG-tagged CSNK2A1 vectors were transfected in HEK293T cells using Polyethylenimine MAX (Polysciences, Warrington, PA, USA) in accordance with the manufacturer’s instructions. After 24 h incubation, cells were lysed with 0.1% NP-40 lysis buffer (500 mM Tris base, 500 mM NaCl, 0.1% NP-40) containing protease inhibitor cocktails (cOmplete Mini EASYpack, Roche Diagnostics, Mannheim, Germany). The lysates for co-immunoprecipitation (IP) assay were incubated with 250 ng anti-DYKDDDK (TransGenic, Fukuoka, Japan, 2H8) and Dynabeads Protein G (Thermo Fisher Scientific) at 4 °C for 1 h. The beads were washed three times with lysis buffer and proteins were eluted with 1X SDS sample buffer. Samples were separated using SDS polyacrylamide gel electrophoresis and analyzed via western blotting with rabbit polyclonal antibodies against HA tag (Roche Diagnostics, 11 867 431 001), DYKDDDK tag (TransGenic) and GFP (GE Healthcare, 27457701).
Results
Clinical features
The clinical features of the four individuals with CSNK2A1 and CSNK2B variants are summarized in Table 1. Two patients with CSNK2A1 variants shared global developmental delays, hypotonia, and intractable seizures, but each exhibited interesting features. Patient 1 showed late-onset and less-frequent seizures (Fig. 1a) and exhibited various abnormalities, including facial dysmorphisms (Supple. Fig S1), brain abnormalities (Fig. 2a, b), short stature, congenital biliary dilatation, and pharyngeal dysphagia. This patient also showed distinct easy fatigability and muscle weakness. These manifestations progressively worsened and led to gait difficulty and respiratory failure. Subsequently, the patient needed a wheelchair for moving and needed continuous mechanical ventilation support. Meanwhile, patient 2 presented a severer course, with seizures resembling those with Dravet syndrome. This patient began to show seizures at the early infantile period, with acute encephalopathy resulting in brain death at the age of 1 year and 7 months. He showed no dysmorphic features or brain abnormalities. These two patients suggested that some cases of CSNK2A1-related disorders could show progressive and serious clinical conditions.

Electroencephalogram (EEG) of patient 1 a and patient 3 b–d. a EEG of patient 1 at 12 years. Course of EEG of patient 3 at 2 months b and 9 years c–d. b Frequent right parietal spikes were present during sleep. c Frequent semi-rhythmic generalized bifrontally predominant 2 c/s spike-and-wave complex were seen during wakefulness. d Occasional generalized 10-Hz fast rhythms were seen during sleep

Brain images of patient 1 a, b and patient 3 c–f. Axial T2-weighted a and sagittal T1-weighted b brain magnetic resonance imaging (MRI) images of patient 1 at 12 years show volumetric loss in cerebral white matter around the posterior horn of the lateral ventricular. Coronal c and sagittal d 99mTc-ethyl cysteinate dimer (ECD) brain single-photon emission computed tomography of patient 3 at 8 months show decreased radiotracer uptake in the cerebellum. Cerebellar atrophy of the vermis and hemisphere were observed in coronal FLAIR e and sagittal T1-weighted brain MRI images f at 14 years
Clinical outcomes of our two cases with CSNK2B variants were both severe. They had seizures within 2 months of age, which were refractory to multiple antiepileptic drugs. Patient 3 showed facial clonic seizures evolving into generalized tonic-clonic seizures at 2 months of age, which developed into Lennox-Gastaut syndrome at 9 years. Patient 4 showed focal seizures on the 5th day after birth; these seizures currently occur 2 or 3 times per day. Severe psychomotor impairment was recognized. Both patients 3 and 4 are bedridden and not able speak any meaningful words. Brain image analysis showed cerebellar atrophy and mega cisterna magna in patients 3 (Fig. 2e, f) and 4, respectively. Patient 3 was diagnosed with precocious puberty at 4 years. Patient 4 was frequently hospitalized due to acute gastroenteritis and pneumonia caused by gastroesophageal reflux disease. Case reports are available in supplementary note.
Identification of de novo CSNK2A1 and CSNK2B variants
Using trio-based WES data of patients 1 and 3, we searched variants consistent with autosomal dominant (including de novo) and autosomal or X-linked recessive models. We found three candidate de novo variants, ATAD2B (NM_017552.2:c.1699G>A, p.Glu567Lys), TOPORS (NM_005802.4:c.298G>A, p.Asp100Asn), and CSNK2A1 (NM_177559.2:c.593A>G, p.(Lys198Arg) in patient 1 (Table 2 and S1). These variants were predicted to be deleterious and were absent in 575 in-house control exomes, public databases, including dbSNP137 data, 1KJPN, the 1000 Genomes Project, and ExAC. We evaluated the pathogenicity of these variants based on the ACMG variant classification guideline [39]. Both variants in ATAD2B and TOPORS were classified as likely being pathogenic, and a variant in CSNK2A1 was classified as pathogenic because the c.593A>G, p.(Lys198Arg) variant in CSNK2A1 had been reported in patients with Okur-Chung neurodevelopmental syndrome [23, 24, 26]. Previous studies suggested missense variants in TOPORS cause autosomal dominant retinitis pigmentosa [40]. Previous patients with TOPORS variants showed no neurodevelopmental disorders, and patient 1 exhibited no manifestations of retinal degenerations. ATAD2B encodes a nuclear protein belonging to the AAA ATPase family and may play a role in neuronal differentiation and tumor progression, though its biological functions and pathogenicity of variants were still uncertain [41]. Considering these findings, we concluded that a de novo CSNK2A1 variant was the most-plausible causative variant in this patient. On the other hand, we found only one candidate de novo variant, CSNK2B (NM_001320.5:c.533_534insGT, p.Pro179Tyrfs*49), in patient 3 (Table 2 and S1), and this variant was classified as pathogenic based on the ACMG guideline. We then searched for possible pathogenic CSNK2A1 and CSNK2B variants using case-only WES data of 893 individuals and identified a novel CSNK2A1 variant (NM_177559.2:c.571C>T, p.Arg191*) and a novel CSNK2B variant (NM_001320.5:c.494A>G, p.His165Arg) in patients 2 and 4, respectively (Table 2). In patients 2 and 4, we also searched for other candidate variants in known causative genes for epileptic encephalopathies, including Dravet syndrome, but could not find any possible pathogenic variants. Altered Arg191 in CSNK2A1 and His165 in CSNK2B residues are evolutionarily highly conserved (Fig. 3a, b). In accordance with the ACMG guideline, CSNK2A1 (c.571C>T) and CSNK2B (c.494A>G) variants were classified as pathogenic and likely pathogenic, respectively. Sanger sequencing confirmed that all four variants occurred de novo (Supple. Fig. S2). In light of these findings, we concluded that their clinical courses were very likely to be related to the CSNK2A1 and CSNK2B variants. The biological parentage of all families was confirmed by analyzing 12 microsatellite markers (data not shown).

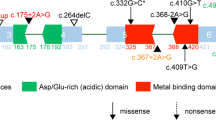
Locations of de novo variants in CSNK2A1 a and CSNK2B b are shown with schematic protein structure. Arrows depicted above are previously reported variants. Four variants found in our cases are shown below. All missense variants occurred at evolutionarily conserved amino acids. Multiple amino acid sequences of CSNK2A1 and CSNK2B proteins were aligned with tools available on the CLUSTALW web site (http://www.genome.jp/tools/clustalw/). a The green box indicates protein kinase domain annotated by Pfam. The protein kinase domain harbors the ATP/GTP binding loop (45–53 aa, pink), basic cluster (68–80 aa, red), active site (156 aa, black) and activation segment (175–201 aa, orange). b The blue box indicates CK_II_beta domain annotated by Pfam. The N-terminal autophosphorylation site (2–6 aa, purple) is attached to the CK_II_beta domain, which contains an acidic cluster (55–64 aa, yellow) and interaction domain with CK2α subunit (188–193 aa, brown). Protein expression c and interaction d of CSNK2B and CSNK2A1. c Representative western blots using extracts from HEK293T cells transiently expressing CSNK2B. Normalization was performed with GFP as a loading control. d HEK293T cells were co-transfected with WT or mutant HA-tagged CSNK2B and FLAG-tagged CSNK2A1 plasmids. Cell lysates were immunoprecipitated with anti-FLAG antibody beads and then probed with anti-HA for CSNK2B and anti-FLAG for CSNK2A1
Protein stability and binding ability of mutant CSNK2B
To examine the protein stability of mutant CSNK2B, HA-tagged WT, or mutant CSNK2B, along with nuclear-localized enhanced green fluorescent protein was transiently expressed in HEK23T cells. Although the protein level of p.Thr37Tyrfs*5 mutant was extremely low, that of two mutants—p.His165Arg and p.Pro179Tyrfs*49—was equivalent to that of WT (Fig. 3c). This result indicated that truncated CSNK2B protein would be degraded in vivo, but prolonged CSNK2B protein would escape from protein degeneration. Next, using co-IP assay we examined the protein interaction between the CSNK2A1 and WT or mutant CSNK2B proteins. The three independent co-IP experiments indicated that wild type and p.His165Arg mutant CSNK2B showed comparable interaction to the CSNK2A1 protein; however, p.Pro179Tyrfs*49 mutant CSNK2B showed no interaction with the CSNK2A1 (Fig. 3d). These findings suggested that p.Pro179Tyrfs*49 mutant may lack the binding ability with CSNK2A1, which may induce the instability of the protein structure of CK2 holoenzyme.
Discussion
In recent years, 26 patients with de novo CSNK2A1 variants have been described [23,24,25,26, 29]. CK2 has two types catalytic subunits, CK2α, and CK2α′, which are encoded from CSNK2A1 and CSNK2A2, respectively. Although both CSNK2A1 and CSNK2A2 are extremely intolerant of single nucleotide variations (Z-score = 3.89 for CSNK2A1, 3.49 for CSNK2A2) and loss-of-function variants (pLI = 1) [36] in humans, pathogenic variants in CSNK2A2 associated with NDDs has not been identified. Previous animal models showed that CK2α knockout mice were embryonic lethal [42, 43]. Conversely, CK2α′ knockout mice showed normal embryonic development and were viable, but the male mice had oligospermia, which led to male infertility [44]. These mutant mice studies suggest that CSNK2A1 variants may have more severe pathogenicity than CSNK2A2 variants at least in neuronal development.
Including the two patients mentioned here, patients having de novo CSNK2A1 variants showed neurodevelopmental disorders with various congenital anomalies (Table 1 and Suppl. Table S2). The most general findings were developmental delay (28/28), intellectual disabilities (26/28), and hypotonia (20/28). In many cases, they accompanied neurological deficits, including motor disorders (11/28), seizures (8/28), autistic traits (8/28), sleep problems (7/28), and attention deficit hyperactivity disorder (3/28). Observed seizures types were febrile (3/8), atonic (1/8), breath-holding spells (1/8), absence (1/8), myoclonic (1/8), and tonic clonic (1/8). Facial dysmorphisms (13/28), short stature (11/28), extremities abnormalities (10/28), and gastrointestinal disorders (10/28) were sometimes noted. As other physical findings, skeletal disorders (8/28), immunological problems (7/28), cardiac abnormalities (5/28), and skin abnormalities (4/28) were occasionally noted (Table S2).
In our study, some characteristic clinical features—congenital biliary dilatation, easy fatigability, and progressive muscle weakness—were noted in patient 1. One previous case (individual 4, Okur et al., Table S2) also presented easy fatigability [23], so this may be one of the minor findings of CSNK2A1-related disorders. Progressive easy fatigability suggested the possibility of myopathies, such as glycogenosis or myasthenia gravis. Previous studies suggested CK2 interacts and co-localizes with the muscle-specific receptor tyrosine kinase (MuSK) at the neuromuscular junction (NMJ) [45]. CK2 mediates the phosphorylation of serine residues within a specific MuSK epitope, then the signal transduction mediates by MuSK-induced aggregation of the acetylcholine receptor (AChR) at the endplate, leading to increasing of the receptor effect to acetylcholine [46]. The loss-of-CK2 function induced fragmentation of AChR clustering, resulting in disintegration of the muscle endplate; notably, muscle-specific CK2β knockout mice develop a myasthenic phenotype due to impaired muscle endplate structure and function [45, 47]. These findings imply CK2 may play important roles in the muscular nerve transmission mechanism and loss-of-CK2 function may be involved in the pathogenesis of muscle weakness; however, the examination of myopathology and repeatedly stimulated nerve conduction studies in patient 1 showed no evident abnormal findings. The impact of CKα functional disability in the signal transduction at NMJ was still uncertain; therefore, further research is needed to prove the relevance of variants in CSNK2A1 in muscle weakness. Patient 2 incurred the most serious clinical outcome among 28 patients with CSNK2A1 variants. This patient only showed developmental delays and frequent epileptic attacks, and died from acute encephalopathy at 1 year and 7 months. This case suggested that variants in CSNK2A1 could cause early onset epileptic encephalopathies.
Patient 1 had a missense variant c.593A>G, p.Lys198Arg, which was hotspot variant of Okur-Chung neurodevelopmental syndrome. Clinical features of eight patients with p.Lys198Arg variants do not entirely match. These variegated phenotypes might be because CK2 holoenzyme comprises identical (α/α or α′/α′) or non-identical (α/α′) combination of catalytic subunits and its enzyme damaging might be variable, depending on heterozygous or homozygous CKα mutants present in the complex. We also found a novel nonsense variant c.571C>T, p.Arg191* caused lack of C-terminal of CK2α protein in patient 2. These two variants occurred at the activation segment, which is the unique alignment of the protein kinase domain of CK2α. The activation segment is fixed in an active conformation and important for CK2 catalytic activity [20]. The amino acid alterations in this region may cause destabilization of this critical conformation and result in impaired kinase activity [48]. Additionally, the alteration in Asp156 residue, referred to as an active site that plays a key role in catalytic activity, had a dominant-negative effect and induced complete loss-of-CK2α enzyme activity [49]. These findings suggest that variants in CSNK2A1 might have dominant-negative effects on CK2α function, but functional confirmation by molecular biological methods is indispensable for this conclusion.
CK2β proteins form homodimers and each CK2β subunit binds to either CK2α or CK2α’ subunit. CK2β acts as a regulator of CK2 holoenzyme, which contributes to the modulation of the enzyme activity, substrate recognition, and stabilization of the holoenzyme complex [50, 51]. CK2β may be crucial for normal embryonic development and CK2β knockout mice were embryonic lethal [52, 53]. CSNK2B is also extremely intolerant toward single-nucleotide variants (Z-score = 3.60) and loss-of-function variants (pLI = 0.94); therefore, CSNK2B variants may cause neurodevelopmental disorders.
Prior to this study, only three patients with CSNK2B variants had been reported [27, 28] and we added two patients with novel CSNK2B variants. Regarding the phenotypes, patients with CSNK2B variants commonly exhibited developmental delay (5/5), intellectual disability (5/5), intractable seizures (4/5), and facial dysmorphisms (3/5) (Table 1 and Suppl. Table S2). The most frequent seizure type was myoclonic (3/4), while tonic, tonic clonic, and focal seizures were also observed. Brain MRI of four patients was normal, but that of patient 3 showed cerebellar atrophy. One patient showed autistic features. Our two cases showed the most severe developmental delay because both patients are currently bedridden and are unable to speak any meaningful words. The number of cases with CSNK2B variants are much smaller than that of CSNK2A1 variants. Therefore, a further study of many CSNK2B cases would be needed for better understanding of the phenotypic spectrum of CSNK2B-related disorders.
Five CSNK2B variants identified to date include two canonical splice site variants, two frameshift variants, and one missense variant (Fig. 3b). Both splice site variants, c.175+2T>G and c.367+2T>C, induced the aberrant exon skipping, which resulted in generating premature stop codons (p.Val25Metfs*13 and p.Leu98Alafs*11, respectively) [27]. Along with a frameshift variant (c.108dup, p.Thr37Thyfs*5) [28], mutant transcripts with these variants are likely subjected to the nonsense-mediated mRNA decay mechanism. Therefore, CSNK2B haploinsufficiency is the pathomechanism in these patients. Our transient expression analysis demonstrated the protein instability of this frameshift mutant and supported this hypothesis (Fig. 3c). Conversely, the mutant transcript with a frameshift variant (c.533_534insGT, p.Pro179Tyrfs*49) could be escaped from the decomposition through nonsense-mediated mRNA decay, and produced the altered and prolonged C terminus of CK2β protein (Fig. 3c). The CK2β tail (Asn181–Arg215) plays key roles in stabilizing the β–β dimer and the β–α contacts [20]. The result of co-IP analysis indicated this prolonged CK2β protein lost the binding ability to CKα protein (Fig. 3d). Therefore, this frameshift variant is likely to cause the instability of the protein structure of CK2 holoenzyme. We also found the missense variant, c.494A>G, p.His165Arg, on an evolutionarily highly conserved residue at CK_II_beta domain (Fig. 3b). Previous studies suggested that the amino acid sequence between Asp155 and Val170 of CK_II_beta domain may have the important role in tightening the β–β interaction [22, 54]; therefore, this amino acid alteration may lead the instability of the β–β dimer formation. We speculate that these two variants (p.His165Arg and p.Pro179Tyrfs*49) may have a dominant-negative effect and impair the CK2 enzyme activities; however, functional analyses are required to prove these hypotheses.
In conclusion, we have identified four patients with CSNK2A1 or CSNK2B variants. Although our study is limited to individuals with early childhood-onset epilepsy, our study expands the phenotypic spectrum of CSNK2A1-related disorders and support the pathogenicity of CSNK2B variants in the patients with neurodevelopmental disorders. Further cases, especially with CSNK2B variants, are needed to delineate the clinical phenotype of CK2-related disorders and genotype–phenotype correlation.
URLs. 1KJPN (https://ijgvd.megabank.tohoku.ac.jp/) 1000 genome (http://www.internationalgenome.org/) ExAC (http://exac.broadinstitute.org) Polyphen-2 (http://genetics.bwh.harvard.edu/pph2/) CADD (http://cadd.gs.washington.edu/) M-CAP (http://bejerano.stanford.edu/mcap/) GERP (http://mendel.stanford.edu/SidowLab/downloads/GERP/index.html) PhastCons (http://compgen.cshl.edu/phast/)
References
Harris JC. New classification for neurodevelopmental disorders in DSM-5. Curr Opin Psychiatry. 2014;27:95–97.
Soden SE, Saunders CJ, Willig LK, Farrow EG, Smith LD, Petrikin JE, et al. Effectiveness of exome and genome sequencing guided by acuity of illness for diagnosis of neurodevelopmental disorders. Sci Transl Med. 2014;6:265ra168.
Miller AR, Masse LC, Shen J, Schiariti V, Roxborough L. Diagnostic status, functional status and complexity among Canadian children with neurodevelopmental disorders and disabilities: a population-based study. Disabil Rehabil. 2013;35:468–78.
Blencowe H, Lee AC, Cousens S, Bahalim A, Narwal R, Zhong N, et al. Preterm birth-associated neurodevelopmental impairment estimates at regional and global levels for 2010. Pediatr Res. 2013;74(Suppl 1):17–34.
Deciphering Developmental Disorders Study. Prevalence and architecture of de novo mutations in developmental disorders. Nature. 2017;542:433–8.
Lelieveld SH, Reijnders MR, Pfundt R, Yntema HG, Kamsteeg EJ, de Vries P, et al. Meta-analysis of 2,104 trios provides support for 10 new genes for intellectual disability. Nat Neurosci. 2016;19:1194–6.
de Ligt J, Willemsen MH, van Bon BW, Kleefstra T, Yntema HG, Kroes T, et al. Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med. 2012;367:1921–9.
Gilissen C, Hehir-Kwa JY, Thung DT, van de Vorst M, van Bon BW, Willemsen MH, et al. Genome sequencing identifies major causes of severe intellectual disability. Nature. 2014;511:344–7.
Iossifov I, O’Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature. 2014;515:216–21.
St-Denis NA, Litchfield DW. Protein kinase CK2 in health and disease: from birth to death: the role of protein kinase CK2 in the regulation of cell proliferation and survival. Cell Mol Life Sci. 2009;66:1817–29.
Guerra B, Issinger OG. Protein kinase CK2 and its role in cellular proliferation, development and pathology. Electrophoresis. 1999;20:391–408.
Loizou JI, El-Khamisy SF, Zlatanou A, Moore DJ, Chan DW, Qin J, et al. The protein kinase CK2 facilitates repair of chromosomal DNA single-strand breaks. Cell. 2004;117:17–28.
Gotz C, Montenarh M. Protein kinase CK2 in development and differentiation. Biomed Rep. 2017;6:127–33.
Ghavidel A, Schultz MC. TATA binding protein-associated CK2 transduces DNA damage signals to the RNA polymerase III transcriptional machinery. Cell. 2001;106:575–84.
Seldin DC, Landesman-Bollag E, Farago M, Currier N, Lou D, Dominguez I. CK2 as a positive regulator of Wnt signalling and tumourigenesis. Mol Cell Biochem. 2005;274:63–67.
Trembley JH, Wang G, Unger G, Slaton J, Ahmed K. Protein kinase CK2 in health and disease: CK2: a key player in cancer biology. Cell Mol Life Sci. 2009;66:1858–67.
Ahmad KA, Wang G, Unger G, Slaton J, Ahmed K. Protein kinase CK2--a key suppressor of apoptosis. Adv Enzym Regul. 2008;48:179–87.
Bodenbach L, Fauss J, Robitzki A, Krehan A, Lorenz P, Lozeman FJ, et al. Recombinant human casein kinase II. A study with the complete set of subunits (alpha, alpha’ and beta), site-directed autophosphorylation mutants and a bicistronically expressed holoenzyme. Eur J Biochem. 1994;220:263–73.
Duncan JS, Litchfield DW. Too much of a good thing: the role of protein kinase CK2 in tumorigenesis and prospects for therapeutic inhibition of CK2. Biochim Biophys Acta. 2008;1784:33–47.
Niefind K, Guerra B, Ermakowa I, Issinger OG. Crystal structure of human protein kinase CK2: insights into basic properties of the CK2 holoenzyme. EMBO J. 2001;20:5320–31.
Litchfield DW. Protein kinase CK2: structure, regulation and role in cellular decisions of life and death. Biochem J. 2003;369:1–15.
Pinna LA. Protein kinase CK2. Int J Biochem Cell Biol. 1997;29:551–4.
Okur V, Cho MT, Henderson L, Retterer K, Schneider M, Sattler S, et al. De novo mutations in CSNK2A1 are associated with neurodevelopmental abnormalities and dysmorphic features. Hum Genet. 2016;135:699–705.
Owen CI, Bowden R, Parker MJ, Patterson J, Patterson J, Price S et al. Extending the phenotype associated with the CSNK2A1-related Okur-Chung syndrome-A clinical study of 11 individuals. Am J Med Genet A. 2018;176:1108–14.
Trinh J, Huning I, Budler N, Hingst V, Lohmann K, Gillessen-Kaesbach G. A novel de novo mutation in CSNK2A1: reinforcing the link to neurodevelopmental abnormalities and dysmorphic features. J Hum Genet. 2017;62:1005–6.
Chiu ATG, Pei SLC, Mak CCY, Leung GKC, Yu MHC, Lee SL et al. Okur-Chung neurodevelopmental syndrome: eight additional cases with implications on phenotype and genotype expansion. Clin Genet. 2018;93:880–90.
Poirier K, Hubert L, Viot G, Rio M, Billuart P, Besmond C, et al. CSNK2B splice site mutations in patients cause intellectual disability with or without myoclonic epilepsy. Hum Mutat. 2017;38:932–41.
Sakaguchi Y, Uehara T, Suzuki H, Kosaki K, Takenouchi T. Truncating mutation in CSNK2B and myoclonic epilepsy. Hum Mutat. 2017;38:1611–2.
Akahira-Azuma M, Tsurusaki Y, Enomoto Y, Mitsui J, Kurosawa K. Refining the clinical phenotype of Okur-Chung neurodevelopmental syndrome. Hum Genome Var. 2018;5:18011.
Saitsu H, Nishimura T, Muramatsu K, Kodera H, Kumada S, Sugai K, et al. De novo mutations in the autophagy gene WDR45 cause static encephalopathy of childhood with neurodegeneration in adulthood. Nat Genet. 2013;45:445–9, 449e441.
Fukai R, Saitsu H, Tsurusaki Y, Sakai Y, Haginoya K, Takahashi K, et al. De novo KCNH1 mutations in four patients with syndromic developmental delay, hypotonia and seizures. J Hum Genet. 2016;61:381–7.
Hiraide T, Nakashima M, Yamoto K, Fukuda T, Kato M, Ikeda H, et al. De novo variants in SETD1B are associated with intellectual disability, epilepsy and autism. Hum Genet. 2018;137:95–104.
Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164.
Nagasaki M, Yasuda J, Katsuoka F, Nariai N, Kojima K, Kawai Y, et al. Rare variant discovery by deep whole-genome sequencing of 1,070 Japanese individuals. Nat Commun. 2015;6:8018.
Genomes Project C, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, et al. A global reference for human genetic variation. Nature. 2015;526:68–74.
Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–91.
Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46:310–5.
Jagadeesh KA, Wenger AM, Berger MJ, Guturu H, Stenson PD, Cooper DN, et al. M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat Genet. 2016;48:1581–6.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Chakarova CF, Papaioannou MG, Khanna H, Lopez I, Waseem N, Shah A, et al. Mutations in TOPORS cause autosomal dominant retinitis pigmentosa with perivascular retinal pigment epithelium atrophy. Am J Hum Genet. 2007;81:1098–103.
Leachman NT, Brellier F, Ferralli J, Chiquet-Ehrismann R, Tucker RP. ATAD2B is a phylogenetically conserved nuclear protein expressed during neuronal differentiation and tumorigenesis. Dev Growth Differ. 2010;52:747–55.
Dominguez I, Degano IR, Chea K, Cha J, Toselli P, Seldin DC. CK2alpha is essential for embryonic morphogenesis. Mol Cell Biochem. 2011;356:209–16.
Lou DY, Dominguez I, Toselli P, Landesman-Bollag E, O’Brien C, Seldin DC. The alpha catalytic subunit of protein kinase CK2 is required for mouse embryonic development. Mol Cell Biol. 2008;28:131–9.
Xu X, Toselli PA, Russell LD, Seldin DC. Globozoospermia in mice lacking the casein kinase II alpha’ catalytic subunit. Nat Genet. 1999;23:118–21.
Cheusova T, Khan MA, Schubert SW, Gavin AC, Buchou T, Jacob G, et al. Casein kinase 2-dependent serine phosphorylation of MuSK regulates acetylcholine receptor aggregation at the neuromuscular junction. Genes Dev. 2006;20:1800–16.
Herrmann D, Straubinger M, Hashemolhosseini S. Protein kinase CK2 interacts at the neuromuscular synapse with Rapsyn, Rac1, 14-3-3gamma, and Dok-7 proteins and phosphorylates the latter two. J Biol Chem. 2015;290:22370–84.
Eiber N, Simeone L, Hashemolhosseini S. Ablation of protein kinase CK2beta in skeletal muscle fibers interferes with their oxidative capacity. Pharmaceuticals. 2017;10:13.
Kuntamalla PP, Kunttas-Tatli E, Karandikar U, Bishop CP, Bidwai AP. Drosophila protein kinase CK2 is rendered temperature-sensitive by mutations of highly conserved residues flanking the activation segment. Mol Cell Biochem. 2009;323:49–60.
Cosmelli D, Antonelli M, Allende CC, Allende JE. An inactive mutant of the alpha subunit of protein kinase CK2 that traps the regulatory CK2beta subunit. FEBS Lett. 1997;410:391–6.
Kusk M, Ahmed R, Thomsen B, Bendixen C, Issinger OG, Boldyreff B. Interactions of protein kinase CK2beta subunit within the holoenzyme and with other proteins. Mol Cell Biochem. 1999;191:51–58.
Raaf J, Issinger OG, Niefind K. First inactive conformation of CK2 alpha, the catalytic subunit of protein kinase CK2. J Mol Biol. 2009;386:1212–21.
Blond O, Jensen HH, Buchou T, Cochet C, Issinger OG, Boldyreff B. Knocking out the regulatory beta subunit of protein kinase CK2 in mice: gene dosage effects in ES cells and embryos. Mol Cell Biochem. 2005;274:31–37.
Buchou T, Vernet M, Blond O, Jensen HH, Pointu H, Olsen BB, et al. Disruption of the regulatory beta subunit of protein kinase CK2 in mice leads to a cell-autonomous defect and early embryonic lethality. Mol Cell Biol. 2003;23:908–15.
Marin O, Meggio F, Sarno S, Pinna LA. Physical dissection of the structural elements responsible for regulatory properties and intersubunit interactions of protein kinase CK2 beta-subunit. Biochemistry. 1997;36:7192–8.
Acknowledgements
We would like to thank the patients’ families for participating in this work. This study was supported by grants for: Research on Measures for Intractable Diseases from Ministry of Health, Labour and Welfare of Japan; Comprehensive Research on Disability Health and Welfare, the Strategic Research Program for Brain Science; Initiative on Rare and Undiagnosed Diseases in Pediatrics from the Japan Agency for Medical Research and Development; Grant-in-Aid for Scientific Research (A)(17H01539), (B)(16H05160), and (C)(15K10367) from the Japan Society for the Promotion of Science.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Nakashima, M., Tohyama, J., Nakagawa, E. et al. Identification of de novo CSNK2A1 and CSNK2B variants in cases of global developmental delay with seizures. J Hum Genet 64, 313–322 (2019). https://doi.org/10.1038/s10038-018-0559-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s10038-018-0559-z
This article is cited by
-
Protein kinase CK2 phosphorylates a conserved motif in the Notch effector E(spl)-Mγ
Molecular and Cellular Biochemistry (2023)
-
Two different presentations of de novo variants of CSNK2B: two case reports
Journal of Medical Case Reports (2022)
-
ACAN biallelic variants in a girl with severe idiopathic short stature
Journal of Human Genetics (2022)
-
Six years’ accomplishment of the Initiative on Rare and Undiagnosed Diseases: nationwide project in Japan to discover causes, mechanisms, and cures
Journal of Human Genetics (2022)
-
Protein kinase CK2: a potential therapeutic target for diverse human diseases
Signal Transduction and Targeted Therapy (2021)
