
Abstract
Long QT syndrome (LQTS) is a rare inherited arrhythmia disease characterized by a prolonged QT interval on 12-lead electrocardiograms. It is the crucial factor to induce syncope, ventricular fibrillation, and even sudden cardiac death. Previous studies have proved that mutations of ion channels-related genes play an important role in LQTS patients. In this study, we enrolled a Chinese family with LQTS and syncope. With the help of whole-exome sequencing, we identified a novel nonsense mutation (c.439C>T/p.Q147X) of Ring Finger Protein 207 (RNF207) in this family. The novel mutation, resulting in a premature stop codon in exon 4 of the RNF207 gene, co-segregated with the affected individuals. Bioinformatics analysis and real-time PCR further proved that the newly identified mutation might induce nonsense-mediated mRNA decay. In mutation carriers, the level of RNF207 mRNA expression was much lower than controls, which may affect potassium channel KCNH2 and lead to LQTS and syncope. In this research, we reported a rare novel mutation of RNF207 in LQTS and syncope patients which further supports the significant role of RNF207 in potassium channel activation and expanded the spectrum of RNF207 mutations. These data may contribute to the genetic diagnosis and counseling of families with LQTS and syncope.
Similar content being viewed by others

Introduction
Long QT syndrome (LQTS) is one kind of the serious arrhythmia disease. It can induce syncope, ventricular fibrillation, and even sudden cardiac death [1, 2]. In clinic, the typical feature of LQTS is a prolonged QT interval on 12-lead electrocardiograms (ECG) and the prevalence of LQTS is about 0.05% [3]. Generally, the inherited pattern of LQTS is autosomal dominant trait with incomplete penetrance. Previous studies have proved that ~15 genes encoding the ion channel play a crucial role in LQTS [4]. However, Due to crucial genetic heterogeneity and complexity of a diverse genetic architecture, the numbers of LQTS-causing genes are much less than candidate genes [3].
RNF207 locates at chromosome 1p36.31 and encodes RNF207 [5], which plays a crucial role in cardiac repolarization possibly by stabilizing membrane expression of the potassium channel KCNH2 (HERG) [6], a known LQTS gene [7]. In addition, some researches revealed that RNF207 was high expressed in the heart [8]. In neonatal rat cardiomyocytes, knocking down RNF207 can induce a reduction of ATP concentration and mitochondrial dysfunction and plays an important role in the development of heart failure [9]. To date, only one mutation of RNF207 has been identified in the patients with arrhythmia [10].
In this study, we have enrolled a Chinese family with arrhythmia. Whole-exome sequencing was employed to detect the genetic lesion of the affected individuals.
Material and methods
The study protocol was approved by the Review Board of the Second Xiangya Hospital of the Central South University in China, and the study participants gave informed consent.
Subjects
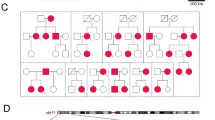
Blood was obtained from 18 family members, including six affected individuals (Fig. 1a). Subjects were reviewed based on their medical records and via B ultrasonic and 12-lead echocardiograms (ECG).

The pedigree and whole-exome sequencing analysis strategies of this family. a Pedigree of the family. Black circles/squares are affected, white are unaffected. Arrow indicates the proband. Three large circles in red represent the three individuals underwent whole-exome sequencing. b Overlapping filter strategy. Asterisks denotes remaining variants for further analysis that are present in two affected members (III-4 and II-6) but not in the normal control (II-1). c Schematic representation of the filter strategies employed in our study
Whole-exome sequencing
Genomic DNA was extracted from peripheral blood lymphocytes of all the family members by using a DNeasy Blood & Tissue Kit (Qiagen, Valencia, CA). The Novogene Bioinformatics Institute (Beijing, China) provided the main part of whole-exome sequencing, including targeted capture, massive parallel sequencing, mapping, variant detection, filtering, and annotation. In this study, we selected the proband (III-4) and two family (II-1 and II-6) members to perform the whole-exome sequencing. The exomes were captured using Agilent SureSelect Human All Exon V6 kits, and the platform of high-throughput sequencing was performed in Illumina HiSeq X-10. The strategies of data filtering are as described previously [11, 12]: (1) variants in the 1000 Genomes project, YH Genomes, dbSNP132 and the NovoDb in-house control with MAF > 0.01 were excluded; (2) Variants shared by two affected family members (III-4 and II-6), but not present in the unaffected normal control (II-1) were remained; (3) Co-segregation analysis combined with bioinformatics analysis validate the damaging causing variants (Fig. 1b, c).
Mutation validation and co-segregation analysis
Sanger sequencing was applied to validate the candidate variants identified in whole-exome sequencing. Co-segregation analysis was conducted in all family members of this study. The primer pairs were designed by Primer 5 (the sequences of primers will be provided upon request), and sequences of the polymerase chain reaction (PCR) products were determined using the ABI 3100 Genetic Analyzer (ABI, Foster City, CA).
RNA extraction and real-time qPCR
Total RNA was extracted by the PureLink® RNA Mini (Thermo Fisher Scientific, #12183025) from patients and healthy controls mononuclear cells. cDNA was synthesized from a total of 1 μg of RNA using the RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific, #K1621) with oligo (dT) primers. Real-time qPCR reactions were carried out in Fast 7500 Real-Time PCR Systems (Applied Biosystems) using Maxima SYBR Green/ROX qPCR Master Mix (2×) (Thermo Fisher Scientific, #K0221). And 2(−△△Ct) was used to compare RNF207 mRNA expression between the affected individuals and the controls. Each assay was performed in five independent tests. The data were analyzed by unpaired two-tailed tests using Graph Pad Prism V.5 software (V.5.0).
Results
Clinical data
We have enrolled a Chinese family with QT prolongation and syncope (Table 1). The proband (III-4), a 34-year-old woman from Hunan province of Central South China, experienced syncope while working in the fields. The ECG record showed a QT prolongation (QTc = 0.51 s) (Fig. 2a). Medical history investigation revealed that the proband has suffered from non-persistent palpitation and chest distress for ~10 years. Family history indicated that her father (II-3) and cousin (III-6) also had the experience of palpitation or syncope. The ECG records of her father (II-3) (QTc = 0.47 s) (Fig. 2b), nephew (IV-3) (QTc = 0.46 s) (Fig. 2c) and aunt (II-6) (QTc = 0.54 s) (Fig. 2d) showed QT internal prolongation (Table 1). Her grandmother (I-2) was died at 51 years old during sleep for unknown reason. The cardiac ultrasound examination showed no obvious structure malformations in the proband.

The clinic data and genetic analysis of this family. The ECG records of III-4 a, II-3 b, IV-3 c, and (II-6) d. e Sanger DNA sequencing chromatogram demonstrates the heterozygosity for a RNF207 nonsense mutation (c.439C>T/p.Q147X). g Analysis of the mutation and protein domains of RNF207. The red arrow represents the novel mutation identified in this study. The blue arrow indicates the previous reported mutation. The Q147 affected amino acid locates in the highly conserved amino acid region in different mammals (from Ensembl). The black arrow shows the Q147 site. f RNA expression of RNF207 in affected individuals and controls. Mean expression ( ± SEM) of RNF207 in affected individual and control measured by real-time qPCR. ***p < 0.01
Genetic analysis
Whole-exome sequencing yielded 9.14 Gb data with 99.6% coverage of target region and 98.7% of target covered over 10 × . In a total of 84,122 variants were present in the proband (Table S1). The identified SNV and indels were analyzed as described in Fig. 1c. Sanger sequencing identified one novel nonsense mutation (c.439C>T/p.Q147X) of RNF207 which co-segregated with the affected individuals of this family (Table 2, Fig. 2e). This mutation, resulting in a premature stop codon in exon 4 of the RNF207 gene, was not found in our 200 local control cohorts [11].
Real-time qPCR analysis
The novel nonsense mutation (c.439C>T/p.Q147X) located in a highly evolutionarily conserved domain (Fig. 2f) and may induce the decay of RNF207 mRNA in the mutation carriers according to nonsense-mediated mRNA decay theory [13, 14]. According to GTEX database, RNF207 can express in whole-white cells, heart tissue and other tissue [15]. We then isolated the mRNA from the mononuclear cells in the patients and healthy family members. We set the mRNA levels of RNF207 in healthy controls as “1”. Real-time qPCR analysis indicated that the mRNA levels of RNF207 were decreased about 53% compared with the healthy controls (p < 0.001) (Fig. 2g).
Discussion
In this study, we employed whole-exome sequencing to detect the genetic lesion of a family with QT prolongation and syncope. A novel nonsense mutation (c.439C>T/p.Q147X) of RNF207 was identified and co-segregated with affected individuals. This mutation was absent in 200 local control cohorts as well as dbSNP132, 1000 genomes and EXAC databases. Functional research further confirmed that the mRNA levels of RNF207 in mutation carriers were obvious lower than healthy controls, indicating the consequence of this mutation. According to HGMD database, there was only one mutation of RNF207 had been reported in arrhythmia patients [10].
RNF207 is an important regulator of cardiac excitation. Previous studies have proved that RNF207 can regulate action potential duration and the repolarizing channel HERG in cardiomyocytes [6]. In addition, some researchers have also found that RNF207 can interact with the voltage-dependent anion channels (VDAC) to regulate the function of mitochondria and energy metabolism of cardiomyocytes [9]. Furthermore, scientists have identified the association between a single nucleotide polymorphism in RNF207 in human genetics and the QT interval [16,17,18]. And the mutation p.G603fsX of RNF207 has also been identified in long QT patients [10] (Fig. 2f). Our study is consistent with previous studies which showed that mutations in RNF207 may lead to long QT and syncope.
RNF207 is consisting of four main domains:[6] a conserved RING domain, a B-box-1 domain, a B-box C-terminal (BBC) domain, and a C-terminal homologous region (Fig. 2f). The conserved RING domain can bind HERG, HSP70, and other chaperones to assist HERG synthesis, folding and to contribute to repolarization reserve [6, 19,20,21,22]. In our case, the nonsense mutation (c.439C>T/p.Q147X) of RNF207 may lead to the reduction of RNF207 expression due to nonsense-mediated mRNA decay. The loss function caused by the novel nonsense mutation, may disturb the synthesis and folding of HERG and result in a reduction repolarization reserve, and, thereby, a long QT and syncope phenotype [6].
In our study, the phenotypes of the affected family members were a little different. III-4 has left axis deviation, II-3 has high voltage, and II-6 has high T wave. These differences may be caused by genetic heterogeneity and environments. For example, the chunky person, the position of heart on the chest, the weight of left and right ventricles all can lead to left axis deviation. In clinic, high voltage is often related to chest wall muscle condition and body fluid condition. And in healthy people, high T wave were also natural condition. However, all the affected members were suffered from LQTS. Our study further confirmed that mutations in RNF207 may lead to LQTS and syncope.
In summary, by using whole-exome sequencing, we have identified a novel nonsense mutation (c.439C>T/p.Q147X) of RNF207 in a Chinese family with long QT and syncope. Real-time qPCR further confirmed that this novel nonsense mutation can lead to functional haploinsufficiency of RNF207 due to nonsense-mediated mRNA decay. Our study reports a rare novel mutation of RNF207 and this is the first nonsense mutation in RNF207. We also expand the spectrum of RNF207 mutations and may contribute to genetic diagnosis and counseling of long QT and syncope.
References
Abrams DJ, Macrae CA. Long QT syndrome. Circulation. 2014;129:1524–9.
Bohnen MS, Peng G, Robey SH, Terrenoire C, Iyer V, Sampson KJ, et al. Molecular pathophysiology of congenital Long QT syndrome. Physiol Rev. 2017;97:89–134.
Giudicessi JR, Wilde AAM & Ackerman MJ. The genetic architecture of long QT syndrome: a critical reappraisal. Trends Cardiovasc Med. 2018,28: 453–64.
Nakano Y, Shimizu W. Genetics of Long-QT syndrome. J Hum Genet. 2016;61:51–5.
Okawa ER, Gotoh T, Manne J, Igarashi J, Fujita T, Silverman KA, et al. Expression and sequence analysis of candidates for the 1p36.31 tumor suppressor gene deleted in neuroblastomas. Oncogene. 2008;27:803–10.
Roder K, Werdich AA, Li W, Liu M, Kim TY, Organ-Darling LE, et al. RING finger protein RNF207, a novel regulator of cardiac excitation. J Biol Chem. 2014;289:33730–40.
Curran ME, Splawski I, Timothy KW, Vincent GM, Green ED, Keating MT. A molecular basis for cardiac arrhythmia: HERG mutations cause Long QT syndrome. Cell. 1995;80:795–803.
Han QY, Wang HX, Liu XH, Guo CX, Hua Q, Yu XH, et al. Circulating E3 ligases are novel and sensitive biomarkers for diagnosis of acute myocardial infarction. Clin Sci. 2015;128:751–60.
Mizushima W, Takahashi H, Watanabe M, Kinugawa S, Matsushima S, Takada S, et al. The novel heart-specific RING finger protein 207 is involved in energy metabolism in cardiomyocytes. J Mol Cell Cardiol. 2016;100:43–53.
Priest JR, Ceresnak SR, Dewey FE, Malloy-Walton LE, Dunn K, Grove ME, et al. Molecular diagnosis of long QT syndrome at 10 days of life by rapid whole genome sequencing. Heart Rhythm. 2014;11:1707–13.
Fan LL, Huang H, Jin JY, Li JJ, Chen YQ, Zhao SP et al. Whole exome sequencing identifies a novel mutation (c.333+2T>C) of TNNI3K in a Chinese family with dilated cardiomyopathy and cardiac conduction disease. Gene. 2018;648:63–7.
Huang H, Chen YQ, Fan LL, Guo S, Li JJ, Jin JY, et al. Whole-exome sequencing identifies a novel mutation of GPD1L (R189X) associated with familial conduction disease and sudden death. J Cell Mol Med. 2018;22:1350–4.
Baker KE, Parker R. Nonsense-mediated mRNA decay: terminating erroneous gene expression. Curr Opin Cell Biol. 2004;16:293–9.
Kurosaki T, Maquat LE. Nonsense-mediated mRNA decay in humans at a glance. J Cell Sci. 2016;129:461–7.
Consortium GT. The Genotype-Tissue Expression (GTEx) project. Nat Genet. 2013;45:580–5.
Newton-Cheh C, Eijgelsheim M, Rice KM, de Bakker PI, Yin X, Estrada K, et al. Common variants at ten loci influence QT interval duration in the QTGEN Study. Nat Genet. 2009;41:399–406.
Pfeufer A, Sanna S, Arking DE, Muller M, Gateva V, Fuchsberger C, et al. Common variants at ten loci modulate the QT interval duration in the QTSCD Study. Nat Genet. 2009;41:407–14.
Noseworthy PA, Havulinna AS, Porthan K, Lahtinen AM, Jula A, Karhunen PJ, et al. Common genetic variants, QT interval, and sudden cardiac death in a Finnish population-based study. Circ Cardiovasc Genet. 2011;4:305–11.
Walker VE, Atanasiu R, Lam H, Shrier A. Co-chaperone FKBP38 promotes HERG trafficking. J Biol Chem. 2007;282:23509–16.
Walker VE, Wong MJ, Atanasiu R, Hantouche C, Young JC, Shrier A. Hsp40 chaperones promote degradation of the HERG potassium channel. J Biol Chem. 2010;285:3319–29.
Badciong JC, Haas AL. MdmX is a RING finger ubiquitin ligase capable of synergistically enhancing Mdm2 ubiquitination. J Biol Chem. 2002;277:49668–75.
Ficker E, Dennis AT, Wang L, Brown AM. Role of the cytosolic chaperones Hsp70 and Hsp90 in maturation of the cardiac potassium channel HERG. Circ Res. 2003;92:e87–100.
Acknowledgements
We thank all subjects for participating in this study. This study was supported by the National Natural Science Foundation of China (81370394 and 81500359).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Fan, LL., Chen, YQ., Huang, H. et al. Exome sequencing identifies a novel nonsense mutation of Ring Finger Protein 207 in a Chinese family with Long QT syndrome and syncope. J Hum Genet 64, 233–238 (2019). https://doi.org/10.1038/s10038-018-0549-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s10038-018-0549-1
