
Abstract
Sinoatrial node dysfunction and deafness (SANDD) syndrome is rare and characterized by a low heart beat and severe-to-profound deafness. Additional features include fatigue, dizziness, and episodic syncope. The sinoatrial node (SAN) drives heart automaticity and continuously regulates heart rate. The CACNA1D gene encoding the Cav1.3 protein expressed in inner hair cells, atria and SAN, induces loss-of-function in channel activity and underlies SANDD. To date, only one variant c.1208_1209insGGG:p.(G403_V404insG) has been reported for SANDD syndrome. We studied five Pakistani families with SANDD and characterized a new missense variant p.(A376V) in CACNA1D in one family, and further characterized the founder variant p.(G403_V404insG) in four additional pedigrees. We show that affected individuals in the four families which segregate p.(G403_V404insG) share a 1.03 MB haplotype on 3p21.1 suggesting they share a common distant ancestor. In conclusion, we identified new and known variants in CACNA1D in five Pakistani families with SANDD. This study is of clinical importance as the CACNA1D founder variant is only observed in families from the Khyber Pakhtunkhwa (KPK) province, in Pakistan. Therefore, screening patients with congenital deafness for SAN dysfunction in this province could ensure adequate follow-up and prevent cardiac failure associated with SAN.
Similar content being viewed by others

Introduction
Deafness is a common sensory impairment which effects almost 278 million people worldwide [1]. Genetic deafness is very heterogeneous and can be syndromic (30% of cases) or non-syndromic (70% of cases). Several syndromes are associated with deafness and heart anomalies, but only two syndromes with deafness and heart problems as the sole features have been described: Jervell and Lange-Nielsen syndrome (JLNS) and Sinoatrial node dysfunction and deafness (SANDD) syndrome (OMIM 614896). KCNQ1 and KCNE1 are associated with the JLNS. Both genes encode potassium channels found in the heart and stria vascularis of the cochlea. SANDD syndrome, characterized by sinoatrial node dysfunction and deafness, was previously reported to be due to an insertion of one glycine residue (c.1208_1209insGGG) in the CACNA1D gene (rs398122827) [2] and no additional variants have been reported.
CACNA1D is an important gene for inner ear cells that encodes the pore-forming α 1D subunit (Cav1.3) of L-type voltage-dependent calcium channels (LTCCs). These channels tightly control Ca2+-dependent glutamate release in the cochlear inner hair cell (IHC) ribbon synapses and are essential for hearing [3]. LTCCs are multisubunit complexes, consisting of alpha-1, alpha-2, beta, and delta subunits. The channel activity is directed by the pore-forming and voltage-sensitive alpha-1D subunit [4]. α1 is a transmembrane domain composed of four homologous domains (D1–D4) each containing six α-helices (S1–S6). S5 and S6 along with linker peptide (S5–S6 linker) form the pore of voltage-dependent calcium channel [5]. These channels have been found to play a role in heart and smooth muscle contraction and in the transmission of auditory information. Cav1.3 is largely absent from the ventricles but is highly expressed in atria and sinoatrial node (SAN) [4]. In the ear, Cav1.3 channels are located at IHC synapses and they mediate Ca2+ influx which is required for transmission of auditory information into the brain via the auditory nerve. LTCCs containing a pore-forming α1D subunit (D-LTCCs) are essential for normal cardiac pacemaking. Loss-of-function of Cav1.3 channels also induces SAN bradycardia in mice [3]. Several cardiac disorders such as sinus bradycardia and tachycardia, sinus arrest, sinus-exit, block and alternating periods of bradycardia, and tachycardia are considered as signs of sinus dysfunction [6]. As there is an important role of Cav1.3 in the heart and central auditory system, it is not surprising that CACNA1D causes a phenotype in both systems [7].
To date, only one variant has been reported for SANDD syndrome which segregated in two separate families. The homozygous CACNA1D c.1208_1209insGGG variant was first described in SANDD individuals in two Pakistani consanguineous families from the KPK province (personal communication with Dr. Shahid Mahmood Baig). It was previously suggested that this variant is possibly a founder mutation [2].
In this study, we describe an additional four families from the KPK province with this same variant in CACNA1D (c.1208_1209insGGG), provide more information on its founder origin, and also characterized a new SANDD syndrome missense variant in CACNA1D in a family also from the same province. If a cardiac dysfunction remains clinically silent in these patients, it can have fatal consequences. Cardiac investigation is therefore highly recommended for individuals with deafness of unknown genetic origin in the KPK province of Pakistan. To avoid the cardiovascular complications, a regular follow-up of individuals with SANDD syndrome is highly recommended. The present study broadens the variant spectrum of SANDD and it also supports that CACNA1D is the causative gene for SANDD.
Methods
Sample collection
This study was approved by the Institutional Review Boards of the Quaid-i-Azam University and the Baylor College of Medicine and Affiliated Hospitals. Written informed consent was obtained from all participating members.
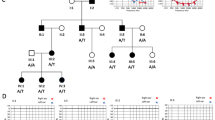
Five unrelated consanguineous families DEM4730, DEM4713, DEM4690, DEM4591, and DEM4318 (Fig. 1) with deafness and cardiac dysfunction were enrolled from different areas of the KPK province. All families displayed an autosomal recessive mode of inheritance for SANDD. Affected members of these families underwent a complete physical examination. Electrocardiograms (ECG) and pure-tone audiometry were performed at a local government hospital.

Pedigree drawings for the families included in this study. a DEM4730, b DEM4713, c DEM4690, d DEM4591, and e DEM4318. Squares represent males and circles represents females; filled symbols represent individuals with SANDD and clear symbols represent the unaffected family members. Double lines show consanguineous marriages. Below each family member of DEM4730 for which a DNA sample is available, their CACNA1D c.1127 C > T genotype is shown. For DEM4713, DEM4690, DEM4591, and DEM4318, the CACNA1D c.1208_1209insGGG genotype is shown for family members from whom a DNA sample is available
Whole-genome genotyping and linkage analysis
Genomic DNA was extracted from peripheral blood using a phenol chloroform procedure [8]. Whole-genome genotyping was performed for all samples using the Illumina Human Core SNP array, which contains ~250,000 single-nucleotide polymorphisms (SNPs), for families DEM4730, DEM4713, and DEM4690, and the Illumina linkage panel bead array, which includes 6000 SNP markers for families DEM4591 and DEM4318. We performed two-point and multipoint linkage analyses using Merlin [9] assuming an autosomal recessive mode of inheritance with complete penetrance, no-phenocopies, and a disease frequency of 0.001. Marker allele frequencies were estimated from the founders and reconstructed founders who were genotyped at the same time while the genetic map positions (cM) were interpolated using the Rutgers Combined Linkage-Physical map. Homozygosity mapping was also performed to analyze the genotype data using Homozygosity Mapper [10].
Exome sequencing
Exome sequencing was performed using a DNA sample from an affected family member from each family: DEM4730 (IV:2); DEM4713 (IV:1); DEM4690 (IV:1); DEM4591 (V:3); and DEM4318 (IV:2) (Fig. 1a–e). Roche NimbleGen SeqCap EZ Human Exome Library preparation kit v.2.0 was used, which selects ~37 Mb of sequence target. Sequencing was performed by 70 bp paired-end sequencing on a HiSeq2500/4000 instrument (Illumina Inc, San Diego, CA, USA). Reads were aligned to the Human genome (hg19/GRC37) using the Burrows–Wheeler algorithm [11] and duplicates were removed with Picard (GATKIndelRealigner). Single-nucleotides variants (SNVs) and small insertions/deletions (Indels) were called using GATK [12] and annotated using dbNSFP and ANNOVAR to aid in filtering and interpretation [13, 14].
Filtering was performed for each exome and only frameshift, in-frame indels, non-synonymous, start/stop altering, nonsense, splice-site variants with a minor allele frequency (MAF) < 0.005 in every Genome Aggregation Database (gnomAD) population were retained. Variants conservation scores (e.g., PhastCons, GERP) [14] and Combined Annotation Dependent Depletion (CADD) C-scores [15] were assessed prior to testing for segregation within the pedigrees. Homozygous variants meeting these criteria in mapped linkage intervals and regions of homozygosity were given priority.
Sanger sequencing
Segregation of variants of interest were verified with Sanger sequencing in the five consanguineous families. Primers surrounding region of interest were designed using primer3 software [16]. PCR amplified products were purified with the ExoSAP-IT™ PCR Product Cleanup Reagent and sequenced using the BigDye terminator v3.1 cycle sequencing kit (on an ABI 3130 Genetic Analyzer).
Haplotype analysis
A founder haplotype was constructed by comparing the genotypes of affected family members of DEM4713 IV:1, DEM4591 V:3, DEM4690 IV:1, and DEM4318 IV:2 who are carriers of the c.1208_1209insGGG variant using the exome sequence data (Fig. 4).
Three-dimensional modeling
The amino acid sequence of human voltage-dependent L-type calcium channel subunit alpha-1D protein was obtained from the UniProt database [17]. The homology modeling techniques were utilized to construct the three-dimensional (3D) model of CACNA1D from the atomic coordinates of Protein data Bank (PDB) entry 2ZXE using PyMOL program [18]. The UCSF Chimera Program was used for the visualization of protein structure [19].
Results
Clinical presentation
Deafness was congenital and severe (71-95 dB) or profound (>95 dB) in all frequencies for all affected members of the SANDD families (Fig. 2c; Supplementary Fig. 1). Individual IV:2 from family DEM4730 with new missense variant c.1127 C > T; p.(A376V) displayed congenital severe deafness affecting all frequencies. There were no obvious signs of vestibular dysfunction, but he did report the symptoms of fatigue, dizziness, and dyspnea. For this family, DEM4730, we were only able to record the ECG for affected family member IV:2 at the age of 15 years. We were unable to obtain ECG from his affected brother IV:3 because he was deceased at the time of the present study. Individual IV:2 displayed bradycardia in ECG recordings. His ECG waveform clearly mark regular but slow rhythm, narrow QRS complex, and P wave did not regularly precede the QRS complex. The recorded heart beat rate was 48 beats per minute (bpm) calculated from beat to beat R–R interval (Fig. 2b). The control shows a normal ECG waveform and the heart beat rate was 60 bpm (Fig. 2a). Individual IV:1 from family DEM4713 and IV:2 from DEM4690 also showed an abnormal pattern of ECG waveforms with bradycardia (Supplementary Fig. 2). For the affected family members there were no additional symptoms, nor any other abnormalities observed.

Clinical presentation of affected individual IV:2 from family DEM4730 with SANDD syndrome. a ECG recordings from a control (60 bpm) and b affected family member (IV:2 from DEM4730) with SANDD syndrome (48bpm) c audiogram for the affected family member IV:2 from DEM4730. Pure-tone audiometry was performed between 250 and 8000 Hz and x represents the results for the left ear and o for the right ear
Genotyping and exome sequencing
Linkage analysis and homozygosity mapping in these families identified several homozygous regions on different chromosomes. A homozygous region on chromosome 3p was shared by all families and the highest LOD score was also observed for each family to this region (Table 1). A DNA sample from a single affected family member per family underwent exome sequencing.
In families DEM4713 (IV:1), DEM4690 (IV:1), DEM4591 (V:3), and DEM4318 (IV:2) the same variant in CACNA1D c.1208_1209insGGG: p.(G403_V404insG) was observed and for DEM4730 a missense variant, c.1127 C > T; p.(A376V) was identified in CACNA1D (NM_000720). DNA samples from all available family members underwent Sanger sequencing and the variants segregated in each respective family (Fig. 1a–e).
The c.1127 C > T; p.(A376V) variant has a CADD C-score of 32 is predicted damaging according to Mutation Taster, and is conserved amongst species (GERP++ RS 5.28 and PhyloP20way 0.852). In addition, the variant is not present in the Greater Middle East (GME) Variome Project that contains 1111 unrelated individuals from the Greater Middle East, including 168 Iranian and Pakistani individuals [20].This variant is present in gnomAD at a very low frequency in the exome data (N = 123,134) (MAF = 1.2 × 10−5) and the frequency in the South Asian exomes (N = 15,391) (MAF = 9.7 × 10−5) (Table 1). The variant was not observed in the homozygous state in gnomAD [21]. Sanger sequencing demonstrated the segregation of the homozygous missense variant (Fig. 3b). This variant affects CACNA1D exon 8B (NM_000720), an alternatively spliced exon that is expressed in the IHCs and SAN cells [2]. The variant has been reported in ClinVar (rs759274321), but no peer-reviewed study has reported the clinical significance of this variant to date. The alanine at amino acid position 376 p.(A376V) is completely conserved among various species and other isoforms of LTCC α1 subunit (Fig. 3g). This amino acid is found in the extracellular topological domain of the α 1D subunit (Cav1.3) of LTCCs (Fig. 3a).

Overview of CACNA1D protein domains and sequence/protein data on variant c.1127 C > T; p.(A376V) a Schematic presentation of the predicted transmembrane helices in CACNA1D (NP_000711.1) (adapted from the results of TMHMM2.0) and depiction of amino acid positions of the new missense variant p.(A376V) in S5–S6 linker of D1 and previously reported variant p.(G403_V404insG), located in the S6 helix of Cav1.3. D1–D4 indicates the four homologous domains of α1 subunit. The box indicates the variant found in this study for SANDD syndrome. The dotted line indicates the location of Trp372, which lies in the S5 helix, and forms a hydrophobic interaction with Val376 in the mutated protein (as depicted by 3D modeling). b Sanger sequencing electrograms and segregation of CACNA1D variant c.1127 C > T; p.(A376V) in family DEM4730. c Predicted three-dimensional structure of the CACNA1D protein. The extracellular domain is colored in cyan and the Ala376 residue is shown by a stick model. d Superimposed structure of template (red) and homology model of CACNA1D (cyan). e Close-up view of the structure showing the wild-type f and mutant protein. g Comparison of LTCC paralog α1 subunit isoforms by Clustal omega (Ensemble ids ENSP00000288139, ENSP00000355192, ENSP00000365441, ENSP00000382563 for Cav1.3, Cav1.1, Cav1.4, and Cav1.2 respectively) (upper panel). Sequence alignment of amino acids across CACNA1D orthologs in various species by Homologene (lower panel), showing that the alanine 376 residue (in green) is conserved in paralogs and across species
Haplotype analysis
Comparison of the genotypes surrounding the CACNA1D gene using exome data revealed that affected family members from DEM4713, DEM4690, DEM4591, and DEM4318 all shared a small 1.03 Mb haplotype from chr3:53326534–54354510 (hg19) at 3p21.1, suggesting that the in-frame variant c.1208_1209insGGG arose on a common founder haplotype (Fig. 4). We confirmed that this variant is a founder mutation.

Haplotype analysis of affected individuals from families with SANDD syndrome that segregate the CACNA1D c.1208_1209insGGG variant. Affected individuals in four families share the same haplotype associated with the c.1208_1209insGGG founder variant (a 1.03 Mb region between 53.32 and 54.35 Mb)
3D modeling
To investigate the possible effect of the p.(A376V) variant, 3D modeling was used to inspect putative structural changes caused by this variant. The residue Ala376 is present in the α-helix of third extracellular domain of protein. In the wild-type protein, the short Ala residue is not involved in any intermolecular interaction with the nearby residues. The variant adds a bulky hydrophobic side chain which contributes the hydrophobic interaction with the nearby residue Trp372 which possibly alters the electrostatic potential compared with wild-type (Fig. 3c–f).
Discussion
SANDD syndrome (OMIM 614896) is caused by homozygous variants in the CACNA1D gene on chromosome 3p21. So far only one variant, c.1208_1209insGGG:p.(G403_V404insG), has been reported in two Pakistani families with SANDD syndrome. Individuals who were homozygous for p.(G403_V404insG) had a pronounced SAN dysfunction with SAN arrhythmia, bradycardia, and severe-to-profound deafness [2]. In this study, we identified four additional families DEM4713, DEM4690, DEM4591, and DEM4318 with the same variant, c.1208_1209insGGG: p.(G403_V404insG). In an additional family DEM4730 we also identified a new missense variant c.1127 C > T:p.(A376V) (rs759274321) in exon 8B (NM_000720) of CACNA1D, which is predicted to be pathogenic. This SANDD variant has been reported as pathogenic in ClinVar but no peer-reviewed study has reported its clinical significance. This variant affects the alanine residue in the third extracellular loop, which is in close proximity to the sixth helical domain of CACNA1D where known variant c.1208_1209insGGG:p.(G403_404insG) is present (Fig. 3a). The third extracellular domain is a pore loop which is present in D1 between the S5 (fifth helical domain) and S6 (sixth helical domain) of the α1 subunit. The pore loop acts as a linker between S5 and S6 (S5–S6 linker) (Fig. 3a) and forms the pore region along with S5 and S6 of the α1 subunit [22]. The S5–S6 linker of D1 has an important role in the conductance of calcium ions across the membrane [23]. 3D protein modeling of variant p.(A376V) shows a change in interaction within the protein due to the substitution of a Ala with Val at position 376 in the third extracellular domain (S5–S6 linker) (Fig. 3c–f). Val376 leads to an additional hydrophobic interaction with Trp372 which is present in S5 (Fig. 3f). This additional hydrophobic interaction could affect the shape of the inner part of the pore and influence the movement of calcium ions across the membrane.
We observed a slow heart beat rate along with severe-to-profound deafness in the SANDD-affected individuals (Fig. 2b, c; Supplementary Figs. 1 and 2). The slow heart rate is possibly due to the defect in the Cav1.3 subunit. It is evident from the previous studies that Cav1.3 channels are involved in heart rate control in humans. Hence, sinoartrial node dysfunction slows down the pacemaking activity of the heart [2, 24]. Patients of SANDD have severe-to-profound deafness and sinus bradycardia. Similarly, homozygous knockout mice for CACNA1D exhibit deafness and heart defects [3]. The pleiotropic role of CACNA1D has already been established [7]. Cav1.3 channels could also be a potential drug target to control heart rate for disease conditions.
A recurrent variant that occurs in a single haplotype of a population can be considered a founder mutation, while a variant that occurs on more than one haplotype is considered a hot-spot mutation [25]. Four unrelated families in our study (DEM4713, DEM4690, DEM4591, and DEM4318) with the same variant c.1208_1209insGGG were recruited from the KPK province of Pakistan. We compared SNVs around the c.1208_1209insGGG variant, which indicated that these four families share the same haplotype for a very small region between chr3:53321631 and 54354510 (1.03 Mb) (Fig. 4). The previously reported families with SANDD syndrome also share the same haplotype associated with the c.1208_1209insGGG founder mutation bounded by SNPs rs720625 and rs1526594 (corresponding to a genomic region of 9.1 Mb) [2] which overlaps with the haplotype observed in our four families.
Last, given this founder mutation was found in families from the same region and has been previously reported [2], this suggests the founder mutation could be used for prenatal diagnosis in pregnancies where the parents are carriers. In addition, we recommend testing for cardiac problems in individuals with congenital hearing loss from the KPK province in Pakistan. This will ensure adequate follow-up and could prevent cardiac failure associated with SAN in the future.
Web resources
ANNOVAR, http://annovar.openbioinformatics.org/
Burrows-Wheeler Aligner, http://bio-bwa.sourceforge.net/
Clinvar, https://www.ncbi.nlm.nih.gov/clinvar/
Combined Annotation Dependent Depletion (CADD), http://cadd.gs.washington.edu/
Clustal omega, https://www.ebi.ac.uk/Tools/msa/clustalo/
dbSNP, https://www.ncbi.nlm.nih.gov/projects/SNP/
Exome Aggregation Consortium (ExAC), http://exac.broadinstitute.org/
gnomAD, http://gnomad.broadinstitute.org/
Genome Analysis Toolkit (GATK), https://software.broadinstitute.org/gatk/
Genomic Evolutionary Rate Profiling (GERP), http://mendel.stanford.edu/SidowLab/downloads/gerp/
Greater Middle East (GME) Variome Project, http://igm.ucsd.edu/gme
Homologene, https://www.ncbi.nlm.nih.gov/homologene/?term = cacna1D
MutationTaster, http://www.mutationtaster.org/
Online Mendelian Inheritance of Man (OMIM), https://www.omim.org/
PhastCons and PhyloP, http://compgen.cshl.edu/phast/
Picard, http://broadinstitute.github.io/picard/
PyMol, https://pymol.org/
TMHMM Server v. 2.0, http://www.cbs.dtu.dk/services/TMHMM/
eXome-Hidden Markov Model (XHMM), https://atgu.mgh.harvard.edu/xhmm/
Uniport, http://www.uniprot.org/
UCSF Chimera, http://www.rbvi.ucsf.edu/chimera
References
Morton CC, Nance WE. Newborn hearing screening—a silent revolution. N Engl J Med. 2006;354:2151–64.
Baig SM, Koschak A, Lieb A, Gebhart M, Dafinger C, Nürnberg G, et al. Loss of Ca(v)1.3 (CACNA1D) function in a human channelopathy with bradycardia and congenital deafness. Nat Neurosci. 2011;14:77–84.
Platzer J, Engel J, Schrott-Fischer A, Stephan K, Bova S, Chen H, et al. Congenital deafness and sinoatrial node dysfunction in mice lacking class D L-type Ca2+ channels. Cell. 2000;102:89–97.
Marger L, Mesirca P, Alig J, Torrente A, Dübel S, Engeland B, et al. Functional roles of Ca v 1.3, Ca v 3.1 and HCN channels in automaticity of mouse atrioventricular cells: insights into the atrioventricular pacemaker mechanism. Channels. 2011;5:251–61.
Catterall WA. Signaling complexes of voltage-gated sodium and calcium channels. Neurosci Lett. 2010;486:107–16.
Benson DW, Wang DW, Dyment M, Knilans TK, Fish FA, Strieper MJ, et al. Congenital sick sinus syndrome caused by recessive mutations in the cardiac sodium channel gene (SCN5A). J Clin Invest. 2003;112:1019–28.
Satheesh SV, Kunert K, Ruttiger L, Zuccotti A, Schonig K, Friauf E, et al. Retrocochlear function of the peripheral deafness gene Cacna1d. Hum Mol Genet. 2012;21:3896–909.
Green MR, Sambrook J. Isolation of high-molecular-weight DNA using organic solvents. Cold Spring Harb Protoc. 2017; 2017:356–9.
Abecasis GR, Cherny SS, Cookson WO, Cardon LR. Merlin—rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet. 2002;30:97–101.
Seelow D, Schuelke M. HomozygosityMapper2012—bridging the gap between homozygosity mapping and deep sequencing. Nucleic Acids Res. 2012;40:W516–20.
Li H, Durbin R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics. 2010;26:589–95.
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–303.
Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164–e164.
Liu X, Jian X, Boerwinkle E. dbNSFP: a lightweight database of human nonsynonymous SNPs and their functional predictions. Hum Mutat. 2011;32:894–9.
Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46:310–5.
Koressaar T, Remm M. Enhancements and modifications of primer design program Primer3. Bioinformatics. 2007;23:1289–91.
Chen C, Huang H, Wu CH. Protein bioinformatics databases and resources. Methods Mol Biol. 2017;1558:3–39.
The PyMOL Molecular Graphics System. Version 2.0. Schrödinger, LLC, New York, NY, USA. 2017.
Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. UCSF Chimera? A visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–12.
Greater Middle East Variome Consortium, Scott EM, Halees A, Itan Y, Spencer EG, He Y, et al. Characterization of Greater Middle Eastern genetic variation for enhanced disease gene discovery. Nat Genet. 2016;48:1071–6.
Exome Aggregation Consortium, Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–91.
Striessnig J, Pinggera A, Kaur G, Bock G, Tuluc P. L-type Ca2+ channels in heart and brain. Wiley Interdiscip Rev Membr Transp Signal. 2014;3:15–38.
Dirksen RT, Nakai J, Gonzalez A, Imoto K, Beam KG. The S5-S6 linker of repeat I is a critical determinant of L-type Ca2 + channel conductance. Biophys J. 1997;73:1402–9.
Mangoni ME, Couette B, Bourinet E, Platzer J, Reimer D, Striessnig J, et al. Functional role of L-type Cav1.3 Ca2+ channels in cardiac pacemaker activity. Proc Natl Acad Sci USA. 2003;100:5543–8.
Cavalli-Sforza LL, Feldman MW. The application of molecular genetic approaches to the study of human evolution. Nat Genet. 2003;33(Suppl):266–75.
Acknowledgements
We would like to thank the families for their participation in this study. This work was supported by the Higher Education Commission of Pakistan (to W.A.) and National Institutes of Health (NIH)-National Institute of Deafness and other Disorders grants R01 DC011651 and R01 DC003594 (to S.M.L). “Genotyping services were provided by the Center for Inherited Disease Research (CIDR). CIDR is fully funded through a federal contract from the National Institutes of Health to The Johns Hopkins University, contract number HHSN268201200008I”. Exome sequencing performed at the University of Washington Center for Mendelian Genomics was funded by the NIH–National Human Genome Research Institute grant UM1 HG006493 (to D.A.N., M.J.B., and S.M.L.).
Supplementary data
The Supplementary information in this paper includes the list of members of the University of Washington Center for Mendelian Genomics (UWCMG) study group and two figures (Supplementary figure 1; Supplementary figure 2).
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
10038_2018_542_MOESM1_ESM.docx
Supplementary data: The Supplementary information in this paper includes the list of members of the University of Washington Center for Mendelian Genomics (UWCMG) study group and two figures (Supplementary figure 1; Supplementary figure 2).
Rights and permissions
About this article

Cite this article
Liaqat, K., Schrauwen, I., Raza, S.I. et al. Identification of CACNA1D variants associated with sinoatrial node dysfunction and deafness in additional Pakistani families reveals a clinical significance. J Hum Genet 64, 153–160 (2019). https://doi.org/10.1038/s10038-018-0542-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s10038-018-0542-8
This article is cited by
-
Pathogenicity of de novo CACNA1D Ca2+ channel variants predicted from sequence co-variation
European Journal of Human Genetics (2024)
-
Molecular genetic landscape of hereditary hearing loss in Pakistan
Human Genetics (2022)
-
Calcium signaling in neurodevelopment and pathophysiology of autism spectrum disorders
Molecular Biology Reports (2022)
-
Zebrafish Larvae Carrying a Splice Variant Mutation in cacna1d: A New Model for Schizophrenia-Like Behaviours?
Molecular Neurobiology (2021)
-
Biophysical classification of a CACNA1D de novo mutation as a high-risk mutation for a severe neurodevelopmental disorder
Molecular Autism (2020)
