
Abstract
Carnitine palmitoyltransferase (CPT) II deficiency is one of the most common forms of mitochondrial fatty acid oxidation disorder. Its clinical phenotypes are classified into the muscle, severe infantile, and lethal neonatal forms. Among Caucasians, the muscle form predominates, and the c.338C > T (p.S113L) variant is detected in most cases, whereas among the Japanese, c.1148T > A (p.F383Y) is the variant allele occurring with the highest frequency and can apparently cause symptoms of the severe infantile form. Newborn screening (NBS) for this potentially fatal disease has not been established. We encountered an infantile case of CPT II deficiency not detected in NBS using C16 and C18:1 concentrations as indices, and therefore we adopted the (C16 + C18:1)/C2 ratio as an alternative primary index. As a result, the disease was diagnosed in nine of 31 NBS-positive subjects. The values for (C16 + C18:1)/C2 in the affected newborns partly overlapped with those in unaffected ones. Among several other indices proposed previously, C14/C3 has emerged as a more promising index. Based on these findings, nationwide NBS for CPT II deficiency using both (C16 + C18:1)/C2 and C14/C3 as indices was officially approved and started in April 2018. We diagnosed the disease in four young children presenting with symptoms of the muscle form, whose values for the new indices were not elevated. Although it is still difficult to detect all cases of the muscle form of CPT II deficiency in NBS, our system is expected to save many affected children in Japan with the severe infantile form predominating.
Similar content being viewed by others


Introduction
Carnitine palmitoyltransferase (CPT) II is an enzyme bound to the mitochondrial inner membrane. Long-chain fatty acids are transported into the mitochondria as acylcarnitines of the corresponding chain-length via the sequential function of acyl-CoA synthetase, CPT I, and carnitine-acylcarnitine translocase (CACT). These long-chain acylcarnitines, represented by palmitoylcarnitine (C16), are then turned back into acyl-CoA by CPT II to supply substrates for the β-oxidation system (Fig. 1). This pathway was proposed in 1963 [1] and was experimentally supported in 1970 [2]. Further research has revealed that CPT II is ubiquitously expressed and that there are three types of isozymes for CPT I, as follows: IA, the liver type, IB, the muscle type, and IC, the brain type. Detection and chromosomal mapping of the genes responsible for these enzymes are as follows: CPT2 in 1p32 [3, 4], CPT1A in 11q13 [5], CPT1B in 22q13 [6], and CPT1C in 19q13 [7]. Currently, CPT IA and CPT II deficiencies are described as autosomal recessive human diseases.

Schematic representation of the carnitine cycle. CPT carnitine palmitoyltransferase, CACT carnitine-acylcarnitine translocase
Clinical phenotypes
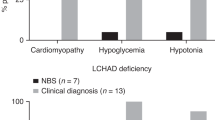
As is characteristic of fatty acid oxidation disorders (FAODs), both CPT IA deficiency and CPT II deficiency can provoke acute metabolic decompensation associated with hypoketotic hypoglycemia. In addition, CPT II deficiency usually shows intermittent myopathic symptoms, including cardiomyopathy in the severest cases, which are not observed in CPT IA deficiency. The first case report of CPT II deficiency appeared in 1973 [8]; the disease was diagnosed in an adult male patient who had exercise-induced muscle cramps and myoglobinuria recurrently over many years. Since then, CPT II deficiency has been clinically classified into the following three phenotypes: (1) an adult-onset muscle form presenting with recurrent rhabdomyolysis in adolescence or later; (2) a severe infantile form which provokes hypoglycemia, Reye-like encephalopathy, and in the worst cases, cardiopulmonary arrest, mainly during infancy and early childhood (the first case was reported by Demaugre et al. [9]); and (3) a lethal neonatal form associated with cardiomyopathy (the first case was reported by Hug et al. [10]). Moreover, increasing data has indicated that not a few patients with the “adult-onset” muscle form have their first episode of myopathic symptoms without hypoglycemia during infancy or early childhood [11].
Genetic backgrounds
Caucasian patients
Numerous studies have reported genotype–phenotype correlation in a considerable number of Caucasian patients in whom c.338C > T (p.S113L) emerged as a common variant at an extremely high frequency [11−22]. On the whole, patients homozygous for p.S113L are expected to present with myopathic symptoms in adolescence and adulthood. However, there have also been sporadic reports of myoglobinuria cases during infancy or early childhood [21, 23, 24]. The compound heterozygous genotype of p.S113L and another null variant is also thought to lead to the muscle form of the disease [18, 25], but there was a case report of cardiac arrest in a 6-year-old girl having this genotype, though her blood glucose level was not documented [26]. On the other hand, there were reports of symptomatic patients who had one allele of p.S113L and were judged to be heterozygous carriers of CPT II deficiency based on an enzyme assay and pedigree analysis [16, 21, 27, 28]. The wide variety of clinical findings associated with the p.S113L variant suggests the presence of additional factors lowering the threshold for the onset of symptoms, such as the coexistence of two “thermolabile polymorphisms”, c.1102G > A (p.V368I) and c.1939A > G (p.M647V), described further below. However, a straightforward interpretation of their effects seems difficult due to the complexity of the clinical information and experimental data reported thus far [29, 30]. A small number of studies of the severe infantile form and the lethal neonatal form of CPT II deficiency in which the genotypes were documented failed to detect the p.S113L variant allele [29, 31−35]. A similar discussion surrounds another mild variant which occurs with considerable frequency, c.149C > A (p.P50H) [30], although this variant was detected in a case of the severe infantile form in combination with a frame-shift variant [31].
Japanese patients
The rarity of the p.S113L variant allele in Japanese patients constitutes one of the significant differences from the Caucasian population. The first Japanese case report of a patient homozygous for p.S113L, published in 2016, described an adult male who experienced recurrent rhabdomyolysis since his teenage years [36]. Currently, c.1148T > A (p.F383Y) is recognized as the variant having the highest frequency among Japanese patients [37−44]. Previous reports described three symptomatic patients homozygous for p.F383Y; the clinical phenotype of these patients was the severe infantile form and one of them died during a Reye-like episode [37, 39]. The phenotype of the five compound heterozygous patients harboring p.F383Y and another variant was either the severe infantile form (four cases including a case of sudden death) [37, 41, 42, 44] or the muscle form (one case) [44]. Table 1 summarizes the information about these cases, excluding the cases cited in refs. [42−44]. Table 2 summarizes the cases described in refs. [42, 44, 45] (S-01 to S-06), and recent cases we encountered as well (S-07 to S-14). The diagnosis of CPT II deficiency was made in 11 of 14 symptomatic patients (S-01 to S-11) based on an assay of CPT II activity in the lysates of lymphocytes and/or the FAO capability of intact lymphocytes; this assessment was confirmed by genetic analysis.
Polymorphisms of thermal instability
Three kinds of single nucleotide substitutions in CPT2 have been described as “thermolabile polymorphisms”, as follows: c.1055T > G (p.F352C), c.1102G > A (p.V368I), and c.1939A > G (p.M647V). According to the ExAC database, the frequencies of the minor alleles are 0.4841 for p.V368I, 0.1620 for p.M647V, and 0.02184 for p.F352C. However, as previous reports indicated, p.F352C seems to be specific to the Japanese and perhaps other East Asian populations, whereas p.M647V seems to be specific to the Caucasian population and p.V368I is apparently shared by both groups [37, 46, 47]. Haplotype analysis of 50 healthy Japanese subjects revealed that the frequencies of p.C352, p.I368, and p.V647 were 0.21, 0.70, and 0.04, respectively [37]. In an in vitro expression experiment, the enzymatic activity of p.F352C-CPT II was found to be 58.9% that of the wild-type enzyme, but p.V368I-CPT II and p.M647V-CPT II showed a relative activity as high as 94.6% and 85.7%, respectively. Apart from their own enzymatic activity, these polymorphisms further impaired the activity of other CPT II variants when one (or more) of them lay on the same allele [48]. In addition, co-expression of p.F352C-V368I-V605L-CPT II and the wild-type enzyme resulted in lower activity than expected for the wild-type, suggesting a dominant-negative effect of these polymorphisms [48]. From the clinical perspective, the frequencies of p.F352C and p.V368I were reportedly higher in Japanese patients presenting with influenza-associated encephalopathy (IAE) than in healthy control subjects, suggesting that they were risk factors for IAE and a possible reason for its higher incidence in the Japanese than in the Caucasian population [47−50].
Among our symptomatic patients, four had the same variant, p.S122F, in one allele (S-11 to S-14 in Table 2). Patient S-11 presented with rhabdomyolysis at age 13 years and received the diagnosis of CPT II deficiency based on his genotype of p.[S122F];[P504L] and low CPT II activity (14.7%). In comparison, patients S-12 to 14 had p.S122F in one allele without any other allelic variants, and their CPT II activities were 45.2%, 50.5%, and 48.7%, respectively. Despite these results indicating that they were heterozygous carriers of CPT II deficiency, the patients presented clinical symptoms possibly related to the disease. Patient S-12 presented with acute encephalopathy at age 9 months, although neither hypoglycemia nor elevated serum CK was observed. Patient S-13 presented with elevated serum CK (up to 3,700IU/L) without hypoglycemia during a febrile episode at age 1 year. Patient S-14 presented with rhabdomyolysis (maximum serum CK value: 13,000IU/L) during an episode of acute gastroenteritis at age 9 months not associated with hypoglycemic symptoms, although the blood glucose level was not measured. With regard to the thermolabile polymorphisms, S-12 had p.F352C and p.V368I on the other allele; S-13 had neither; S-14 was heterozygous for both, but the haplotype was not analyzed. Taken together, the effect of p.F352C and p.V368I on the function of CPT II in these three patients was vague and difficult to evaluate. Further accumulation of data on the correlation between the clinico-biochemical phenotype and genotype, including the haplotype of the thermolabile polymorphisms, is required to clarify their significance.
Newborn screening
On the whole, FAODs are regarded as good targets for newborn screening (NBS) because affected children usually appear to be healthy, and the onset of catastrophic symptoms can be prevented by frequent feeding and, during sick days, by intravenous administration of glucose. In the 1990s, tandem mass spectrometry (MS/MS)-based acylcarnitine analysis was successfully applied to newborn dried blood specimens (DBSs), enabling NBS for FAODs. However, although CPT II deficiency reportedly has the second highest frequency among FAODS in the Caucasian population [51], it is listed as a primary target disease in NBS in a limited number of countries [52, 53], probably due to the possibility of overlooking the vast majority of patients harboring p.S113L [54]. In comparison, a nationwide Japanese survey of symptomatic FAOD cases diagnosed or reported between 1985 and 2000 revealed that CPT II deficiency occurred in 17 of 64 patients (26.6%), indicating that this disease occurred with the highest frequency. Four of the 17 patients exhibited the severe infantile form [55].
CPT II deficiency was included in a pilot study of MS/MS-based NBS, which was conducted from 2004 to 2012 in several areas of Japan using C16 and C18:1 as indices. For NBS, DBSs were generally collected on postnatal day 4 or 5. The new screening system diagnosed CPT II deficiency in seven of 1,740,387 newborns [44]. The frequency of 1/248,627 was lower than expected in comparison with medium-chain acyl-CoA dehydrogenase (MCAD) deficiency (1/108,333) and very-long-chain acyl-CoA dehydrogenase (VLCAD) deficiency (1/162,499). We incidentally encountered a case of an infant with CPT II deficiency who presented with acute hypoglycemic encephalopathy but had passed NBS [42].
Based on the acylcarnitine profile of the newborn DBS from this false-negative patient (S-01) and a previous report demonstrating that (C16 + C18:1)/C2 in the serum or plasma can be a sensitive index for high-risk screening of symptomatic patients [56], we adopted (C16 + C18:1)/C2 and C16 as alternative indices for NBS [44]. The data on the NBS indices and the results of the confirmatory tests are summarized in Table 3 (cited from ref. [44] with modifications and additional data). Based on CPT II activity in the lysates of lymphocytes and/or the FAO capability of intact lymphocytes, the disease was diagnosed in 9 of 31 NBS-positive subjects (N-01 to N-09). Homozygous or compound heterozygous variants of the CPT2 gene were detected in all nine patients. As with symptomatic Japanese patients, the variant occurring with the highest frequency among the NBS-positive patients was p.F383Y, which was detected in seven alleles in six patients from five families. Seven newborns showing mild impairment of CPT II activity and/or FAO capability were presumed to be heterozygous carriers, which was later genetically shown to be the case for two of the patients (N-10 and N-11). The other fifteen newborns showed normal levels of CPT II activity and/or FAO capability.
According to our data on NBS conducted in Japan, the (C16 + C18:1)/C2 values in affected newborns partly overlapped with those in unaffected ones. In order to reduce the false-positive rate, we sought new candidates for the NBS index among various long-chain acylcarnitines, such as C16, C16-OH, C18, C18:1, C18-OH, and their ratios to C2 or C3, and found C14/C3 to be the most promising [44]. Based on these data, CPT II deficiency was finally included among the primary target diseases in July 2017, and the nationwide NBS for CPT II deficiency using (C16 + C18:1)/C2 and C14/C3 as indices (both cutoffs 99.9 percentile) began in April 2018. Figure 2 showing a dual axis graph of (C16 + C18:1)/C2 and C14/C3 in newborn DBS, indicates that C14/C3 is superior to (C16 + C18:1)/C2 when discriminating between affected and unaffected newborns. Among recent cases, however, there were two false-positive newborns with a high C14/C3 ratio overlapping the values of some of the affected ones. The false-positive results were evidently caused by very low levels of C3 rather than by elevated C14. Although a certain level of false-positivity appears to be unavoidable using the current NBS system, the serum concentration of C16 and C18:1 can enable a much clearer differentiation between affected and unaffected newborns (Fig. 3).

Distribution of (C16 + C18:1)/C2 and C14/C3 in newborn dried blood specimens. Black circle Patients (N-01 to 09 except N-04), Grey square Heterozygous carriers (N-10 and N-11), Grey triangle Suspected carriers, Crossmark False-positive subjects, Black diamond False-negative patients (S-07, S-09, and S-10)

Distribution of C16 and C18:1 in serum of NBS-positive subjects. Black circle Patients (N-01 to 09), Grey square Heterozygous carriers (N-10, N-11), Grey triangle Suspected carriers, Crossmark False-positive subjects
Problems to be solved
We recently diagnosed CPT II deficiency in four young children who presented with rhabdomyolytic symptoms without hypoglycemia and whose values for (C16 + C18:1)/C2 and C14/C3 in newborn DBS were below the cutoff (S-07 to S-10). The distribution of the NBS indices in these patients indicated that detecting the muscle form of CPT II deficiency by NBS is not infallible (Fig. 2). Nevertheless, our system is expected to save many affected children in Japan, where the proportion of the severe infantile form of the disease is apparently higher than that in Western countries. Sampling of dried blood specimens earlier than postnatal day 4 or 5, which is not yet practiced in Japan, may improve the detection of the muscle form of CPT II deficiency.
Among the nine patients identified via NBS in our study, two died suddenly during an acute infectious episode (N-01 and N-04), which we believe could have been prevented by stricter management. Five other patients showed myopathic findings despite early therapy (N-02, N-03, and N-05 to N-07), and one had hypoglycemia (N-06). In order to offer optimal medical management to patients with diseases diagnosed via NBS, it is essential to predict their phenotype accurately using objective indices. For this purpose, the correlation between the levels of marker metabolites in the blood, enzymatic function, genotype, and clinical phenotype must be clarified.
However, the genotypes encountered in the present case series were complicated by the presence of p.F352C and p.V368I, whose effects on enzymatic function are difficult to gauge. To understand how substantial their effects are in vivo, it is necessary to analyze genotypes of the parents to determine the haplotypes of these polymorphisms and other variants in as many cases as possible. Another problem is that accurate evaluation of enzymatic function is reportedly difficult for CPT II. Although a variety of methods have been proposed for measuring CPT II activity using cell lysates, the correlation between the values derived from these methods and disease severity has been questioned [4, 12, 30, 46, 57, 58]. However, the long-chain fatty acid oxidation capability of intact cells was found to correlate well with disease severity [30, 32, 59]. Our data appear to agree with those of previous reports. The distribution of CPT II activity in the lymphocytes of patients presenting with the infantile form of the disease ranged from 6.6% to 13.6% of the average value of normal controls (S-01 and S-02 in Table 2 and N-04 in Table 3), whereas that of the patients presenting with the muscle form of the disease ranged from 2.8% to 18.4% (S-04 to S-11 in Table 2 and N-02 in Table 3). In addition, the data from patient S-10 in Table 2 provides useful information. This female patient had rhabdomyolysis at age 4 years and was identified as a compound heterozygote for p.S113L and p.E645*. Her CPT II activity (8.1%) seemed to be too low for the muscle form of the disease and the p.S113L variant enzyme. On the other hand, her FAO capability was 41.9% for d1C2/d31C16 and 33.3% for d27C14/d31/C16, thus possibly better reflecting her phenotype and genotype. As the number of cases of CPT II deficiency confirmed on the basis of both CPT II activity and FAO capability is small, we must continue to accumulate more data.
In view of the fact that NBS for CPT II deficiency has yet to be adopted internationally, we hope that our initiative will contribute greatly to increasing interest in the methods suggested for detecting this potentially fatal disease.
References
Fritz IB, Yue KT. Long-chain carnitine acyltransferase and the role of acylcarnitine derivatives in the catalytic increase of fatty acid oxidation induced by carnitine. J Lipid Res. 1963;4:279–88.
Yates DW, Garland PB. Carnitine palmitoyltransferase activities (EC 2.3.1.-) of rat liver mitochondria. Biochem J. 1970;119:547–52.
Finocchiaro G, Taroni F, Rocchi M, Liras Martin A, Colombo I, Torri Tarelli G, et al. cDNA cloning, sequence analysis, and chomosomal localization of the gene for human carnitine palmitoyltransferase. Proc Natl Acad Sci USA. 1991;88:661–5.
Gellera C, Verderio E, Floridia G, Finocchiaro G, Montermini L, Cavadini P, et al. Assignment of the human carnitine palmitoyltransferase II gene to chromosome lp32. Genomics. 1994;24:195–7.
Britton CH, Mackey DW, Esser V, Foster DW, Burns DK, Yarnall DP, et al. Fine chromosome mapping of the genes for human liver and muscle carnitine palmitoyltransferase I (CPT1A and CPT1B). Genomics. 1997;40:209–11.
Yamazaki N, Shinohara Y, Shima A, Yamanaka Y, Terada H. Isolation and characterization of cDNA and genomic clones encoding human muscle type carnitine palmitoyltransferase I. Biochim Biophys Acta. 1996;1307:157–61.
Price NT, van der Leij FR, Jackson VN, Corstorphine CG, Thomson R, Sorensen A, et al. A novel brain-expressed protein related to carnitine palmitoyltransferase 1. Genomics. 2002;80:433–42.
Di Mauro S, Di Mauro PM. Muscle carnitine palmitoyltransferase deficiency and myoglobinuria. Science. 1973;182:929–31.
Demaugre F, Bonnefont JP, Colonna M, Cepanec C, Leroux JP, Saudubray JM. Infantile form of carnitine palmitoyltransferase II deficiency with hepatomuscular symptoms and sudden death. Physiopathological approach to carnitine palmitoyltransferase II deficiencies. J Clin Invest. 1991;87:859–64.
Hug G, Bove KE, Soukup S. Lethal neonatal multiorgan deficiency of carnitine palmitoyltransferase II. N Engl J Med. 1991;325:1862–4.
Joshi PR, g M, Zierz S. Carnitine palmitoyltransferase II (CPT II) deficiency: genotype–phenotype analysis of 50 patients. J Neurol Sci. 2014;338:107–11.
Taroni F, Verderio E, Dworzak F, Willems PJ, Cavadini P, DiDonato S. Identification of common mutation in the carnitine palmitoyltransferase II gene in familial recurrent myoglobinuria patients. Nat Genet. 1993;4:314–20.
Zierz S, Engel AG, Olek K. The Ser113Leu mutation in the carnitine palmitoyltransferase II gene in patients with muscle carnitine palmitoyltransferase deficiency. Muscle Nerve. 1994;19:S129.
Kaufmann P, El-Schahawi M, DiMauro S. Carnitine palmitoyltransferase II deficiency: diagnosis by molecular analysis of blood. Mol Cell Biochem. 1997;174:237–9.
Bonnefont JP, Demaugre F, Prip-Buus C, Saudubray JM, Brivet M, Abadi N. Carnitine palmitoyltransferase deficiencies. Mol Genet Metab. 1999;68:424–40.
Taggart RT, Smail D, Apolito C, Vladutiu GD. Novel mutations associated with carnitine palmitoyltransferase II deficiency. Hum Mutat. 1999;13:210–20.
Wieser T, Deschauer M, Olek K, Hermann T, Zierz S. Carnitine palmitoyltransferase II deficiency: molecular and biochemical analysis of 32 patients. Neurology. 2003;60:1351–3.
Deschauer M, Wieser T, Zierz S. Muscle carnitine palmitoyltransferase II deficiency: clinical and molecular genetic features and diagnostic aspects. Arch Neurol. 2005;62:37–41.
Isackson PJ, Bennett MJ, Vladutiu GD. Identification of 16 new disease-causing mutations in the CPT2 gene resulting in carnitine palmitoyltransferase II deficiency. Mol Genet Metab. 2006;89:323–31.
Corti S, Bordoni A, Ronchi D, Musumeci O, Aguennouz M, Toscano A, et al. Clinical features and new molecular findings in carnitine palmitoyltransferase II (CPT II) deficiency. J Neurol Sci. 2008;266:97–103.
Fanin M, Anichini A, Cassandrini D, Fiorillo C, Scapolan S, Minetti C, et al. Allelic and phenotypic heterogeneity in 49 Italian patients with the muscle form of CPT-II deficiency. Clin Genet. 2012;82:232–9.
Lehmann D, Zierz S. Normal protein content but abnormally inhibited enzyme activity in muscle carnitine palmitoyltransferase II deficiency. J Neurol Sci. 2014;339:183–8.
Hurvitz H, Klar A, Korn-Lubetzki I, Wanders RJA, Elpeleg ON. Muscular carnitine palmitoyltransferase II deficiency in infancy. Pediatr Neurol. 2000;22:148–50.
Gempel K, von Praun C, Baumkotter J, Lehnert W, Ensenauer R, Gerbitz KD, et al. “Adult” form of muscular carnitine palmitoyltransferase II deficiency: manifestation in a 2-year-old child. Eur J Pediatr. 2001;160:548–51.
Deschauer M, Wieser T, Schröder R, Zierz S. A novel nonsense mutation (515del4) in muscle carnitine palmitoyltransferase II deficiency. Mol Genet Metab. 2002;75:181–5.
Thuillier L, Sevin C, Demaugre F, Brivet M, Rabier D, Droin V, et al. Genotype/phenotype correlation in carnitine palmitoyltransferase II deficiency: lessons from a compound heterozygous patient. Neuromusc Disord. 2000;10:200–5.
Rafay MF, Murphy EG, McGarry JD, Kaufmann P, DiMauro S, Tein I. Clinical and biochemical heterogeneity in an Italian family with CPT II deficiency due to Ser 113 Leu mutation. Can J Neurol Sci. 2005;32:316–20.
Joshi PR, Deschauer M, Zierz S. Clinically symptomatic heterozygous carnitine palmitoyltransferase II (CPT II) deficiency. Wien Klin Wochenschr. 2012;124:851–4.
Olpin SE, Afifi A, Clark S, Manning NJ, Bonham JR, Dalton A, et al. Mutation and biochemical analysis in carnitine palmitoyltransferase type II (CPT II) deficiency. J Inherit Metab Dis. 2003;26:543–57.
Bonnefont JP, Djouadi F, Prip-Buus C, Gobin S, Munnich A, Bastin J. Carnitine palmitoyltransferases 1 and 2: biochemical, molecular and medical aspects. Mol Asp Med. 2004;25:495–520.
Vladutiu GD, Quackenbush EJ, Hainline BE, Albers S, Smail DS, Bennett MJ. Lethal neonatal and severe late infantile forms of carnitine palmitoyltransferase II deficiency associated with compound heterozygosity for different protein truncation mutations. J Pediatr. 2002;141:734–6.
Thuillier L, Rostane H, Droin V, Demaugre F, Brivet M, Kadhom N, et al. Correlation between genotype, metabolic data, and clinical presentation in carnitine palmitoyltransferase 2 (CPT2) deficiency. Hum Mutat. 2003;21:493–501.
Spiegel R, Shaag A, Gutman A, Korman SH, Saada A, Elpeleg O, et al. Severe infantile type of carnitine palmitoyltransferase II (CPT II) deficiency due to homozygous R503C mutation. J Inherit Metab Dis. 2007;30:266.
Isackson PJ, Bennett MJ, Lichter-Konecki UL, Willis M, Nyhan WL, Sutton VR, et al. CPT2 gene mutations resulting in lethal neonatal or severe infantile carnitine palmitoyltransferase II deficiency. Mol Genet Metab. 2008;94:422–7.
Yahyaoui R, Espinosa MG, Gómez C, Dayaldasani A, Rueda I, Roldán A, et al. Neonatal carnitine palmitoyltransferase II deficiency associated with Dandy–Walker syndrome and sudden death. Mol Genet Metab. 2011;104:414–6.
Shima A, Yasuno T, Yamada K, Yamaguchi M, Kohno R, Yamaguchi S, et al. First Japanese case of carnitine palmitoyltransferase II deficiency with the homozygous point mutation S113L. Intern Med. 2016;55:2659–61.
Wataya K, Akanuma J, Cavadini P, Aoki Y, Kure S, Invernizzi F, et al. Two CPT2 mutations in three Japanese patients with carnitine palmitoyltransferase II deficiency: functional analysis and association with polymorphic haplotypes and two clinical phenotypes. Hum Mutat. 1998;11:377–86.
Kaneoka H, Uesugi N, Moriguchi A, Hirose S, Takayanagi M, Yamaguchi S, et al. Carnitine palmitoyltransferase II deficiency due to a novel gene variant in a patient with rhabdomyolysis and ARF. Am J Kidney Dis. 2005;45:596–602.
Aoki J, Yasuno T, Sugie H, Kido H, Nishino I, Shigematsu Y, et al. A Japanese adult form of CPT II deficiency associated with a homozygous F383Y mutation. Neurology. 2007;69:804–6.
Yasuno T, Kaneoka H, Tokuyasu T, Aoki J, Yoshida S, Takayanagi M, et al. Mutations of carnitine palmitoyltransferase II (CPT II) in Japanese patients with CPT II deficiency. Clin Genet. 2008;73:496–501.
Yamamoto T, Tanaka H, Kobayashi H, Okamura K, Tanaka T, Emoto Y, et al. Retrospective review of Japanese sudden unexpected death in infancy: the importance of metabolic autopsy and expanded newborn screening. Mol Genet Metab. 2011;102:399–406.
Kobayashi Y, Ishikawa N, Tsumura M, Fujii Y, Okada S, Shigematsu Y, et al. Acute severe encephalopathy related to human herpesvirus-6 infection in a patient with carnitine palmitoyltransferase 2 deficiency carrying thermolabile variants. Brain Dev. 2013;35:449–53.
Yamada K, Bo R, Kobayashi H, Hasegawa Y, Ago M, Fukuda S, et al. A newborn case with carnitine palmitoyltransferase II deficiency initially judged as unaffected by acylcarnitine analysis soon after birth. Mol Genet Metab Rep. 2017;11:59–61.
Tajima G, Hara K, Tsumura M, Kagawa R, Okada S, Sakura N, et al. Newborn screening for carnitine palmitoyltransferase II deficiency using (C16+C18:1)/C2: evaluation of additional indices for adequate sensitivity and lower false-positivity. Mol Genet Metab. 2017;122:67–75.
Ikeda N, Maruyama S, Nakano K, Imakiire R, Ninomiya Y, Seki S, et al. A surviving 24-month-old patient with neonatal-onset carnitine palmitoyltransferase II deficiency. Mol Genet Metab Rep. 2017;11:69–71.
Taroni F, Verderio E, Fiorucci S, Cavadini P, Finocchiaro G, Uziel G, et al. Molecular characterization of inherited carnitine palmitoyltransferase II deficiency. Proc Natl Acad Sci USA. 1992;89:8429–33.
Chen Y, Mizuguchi H, Yao D, Ide M, Kuroda Y, Shigematsu Y, et al. Thermolabile phenotype of carnitine palmitoyltransferase II variations as a predisposing factor for influenza-associated encephalopathy. FEBS Lett. 2005;579:2040–4.
Yao D, Mizuguchi H, Yamaguchi M, Yamada H, Chida J, Shikata K, et al. Thermal instability of compound variants of carnitine palmitoyltransferase II and impaired mitochondrial fuel utilization in influenza-associated encephalopathy. Hum Mutat. 2008;29:718–27.
Kubota M, Chida J, Hoshino H, Ozawa H, Koide A, Kashii H, et al. Thermolabile CPT II variants and low blood ATP levels are closely related to severity of acute encephalopathy in Japanese children. Brain Dev. 2012;34:20–27.
Yao M, Cai M, Yao D, Xu X, Yang R, Li Y, et al. Abbreviated half-lives and impaired fuel utilization in carnitine palmitoyltransferase II variant fibroblasts. PLoS ONE. 2015;10:e0119936.
Bennett MJ, Rinaldo P, Strauss AW. Inborn errors of mitochondrial fatty acid oxidation. Crit Rev Clin Lab Sci. 2000;37:1–44.
Watson MS, Mann MY, Lloyd-Puryear MA, Rinaldo P, Howell RR. Executive summary. Genet Med. 2006;8:1S–11S.
Loeber JG, Burgard P, Cornel MC, Rigter T, Weinreich SS, Rupp K, et al. Newborn screening programmes in Europe: arguments and efforts regarding harmonization. Part 1—from blood spot to screening result. J Inherit Metab Dis. 2012;35:603–11.
Edmondson AC, Salant J, Ierardi-Curto LA, Ficicioglu C. Missed newborn screening case of carnitine palmitoyltransferase-II deficiency. JIMD Rep. 2017;33:93–97.
Tamaoki Y, Kimura M, Hasegawa Y, Iga M, Inoue M, Yamaguchi S. A survey of Japanese patients with mitochondrial fatty acid β-oxidation and related disorders as detected from 1985 to 2000. Brain Dev. 2002;24:675–82.
Gempel K, Kiechl S, Hofmann S, Lochmuller H, Kiechl-Kohlendorfer U, Willeit J, et al. Screening for carnitine palmitoyltransferase II deficiency by tandem mass spectrometry. J Inherit Metab Dis. 2002;25:17–27.
Verderio E, Cavadini P, Montermini L, Wang H, Lamantea E, Finocchiaro G, et al. Carnitine palmitoyltransferase II deficiency: structure of the gene and characterisation of two novel disease-causing mutations. Hum Mol Genet. 1995;4:19–29.
Yamamoto S, Abe H, Kongo T, Ogawa A, Ohtake A, Hayashibe H, et al. Two novel gene mutations (glu174-lys, phe383-tyr) causing the hepatic form of carnitine palmitoyltransferase II deficiency. Hum Genet. 1996;98:116–8.
Bonnefont JP, Taroni F, Cavadini P, Cepanec C, Brivet M, Saudubray JM, et al. Molecular analysis of carnitine palmitoyltransferase II deficiency with hepatocardiomuscular expression. Am J Hum Genet. 1996;58:971–8.
Acknowledgements
The authors thank Dr. Miyuki Tsumura, Dr. Reiko Kagawa, and Dr. Satoshi Okada (Department of Pediatrics, Hiroshima University Graduate School of Biomedical & Health Sciences) for helping with the genetic analysis, Dr. Nobuo Sakura (Nursing House for Severe Motor and Intellectual Severities, Suzugamine) for assistance with the enzymatic assay, Dr. Ikue Hata and Dr. Yosuke Shigematsu (Department of Pediatrics, School of Medical Sciences, University of Fukui) for assistance with the acylcarnitine analysis and the measurement of fatty acid oxidation capability, and Dr. Seiji Yamaguchi (Department of Pediatrics, Faculty of Medicine, Shimane University) for managing the pilot study on tandem mass spectrometry-based newborn screening in Japan. The authors also thank Ms. Chiyoko Yoshii, Ms. Chiyomi Morioka, and Ms. Saki Fujihara (Hiroshima City Medical Association Clinical Laboratory) for analyzing the dried blood specimens of newborns in Hiroshima prefecture. This study was supported in part by the Health and Labour Sciences Research Grants for (1) Health Research on Children, Youth and Families (Chief Investigator: Go Tajima) and (2) the Research on Rare and Intractable Diseases (Chief Investigator: Kimitoshi Nakamura), by the Practical Research Project for Rare/Intractable Diseases from the Japan Agency for Medical Research and Development, AMED, Grant Number JP18ek0109276 (Chief Investigator: Toshiyuki Fukao), by a Grant-in-Aid for Young Scientists, No. 18K15663, from the Japan Society for the Promotion of Science (Chief Investigator: Miori Yuasa), and by a grant from The Morinaga Hoshi-Kai Foundation. This study was carried out in part at the Analysis Center of Life Science, Hiroshima University, the Institute for Clinical Research at the National Hospital Organization Kure Medical Center and Chugoku Cancer Center.
Funding
Go Tajima received Health and Labour Sciences Research Grants (Health Research on Children, Youth and Families; Chief Investigator: Go Tajima, and Research on Rare and Intractable Diseases; Chief Investigator: Kimitoshi Nakamura) from the Ministry of Health, Labour and Welfare of Japan, and a Practical Research Project for Rare/Intractable Diseases Grant from the Japan Agency for Medical Research and Development, AMED (Grant number JP18ek0109276; Chief Investigator: Toshiyuki Fukao), and a grant from The Morinaga Hoshi-Kai Foundation. Miori Yuasa received a Grant-in-Aid for Young Scientists, No. 18K15663 from the Japan Society for the Promotion of Science. Keiichi Hara did not receive any funding for this study.
Author contributions
Go Tajima developed the assay of CPT II activity, carried out the enzymatic assays for all cases in this study, managed the entire project, and wrote this paper. Keiichi Hara performed the analysis of the CPT2 and CACT genes. Miori Yuasa analyzed the acylcarnitine profiles and measured fatty acid oxidation capability using tandem mass spectrometry.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethics approval
Approval for the biochemical, enzymatic, and genetic studies was obtained from the ethics committees of the National Center for Child Health and Development, Hiroshima University, University of Fukui, National Hospital Organization Kure Medical Center and Chugoku Cancer Center. All procedures were carried out in accordance with the ethical standards of the relevant committees on human experimentation (institutional and national) and the Helsinki Declaration of 1975 as revised in 2000.
Informed consent
Informed consent was obtained from all families enrolled in the study.
The guarantor for the article
Go Tajima.
Rights and permissions
About this article

Cite this article
Tajima, G., Hara, K. & Yuasa, M. Carnitine palmitoyltransferase II deficiency with a focus on newborn screening. J Hum Genet 64, 87–98 (2019). https://doi.org/10.1038/s10038-018-0530-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s10038-018-0530-z
This article is cited by
-
Carnitine Palmitoyl Transferase Deficiency in a University Immunology Practice
Current Rheumatology Reports (2020)
