
Abstract
Mitochondrial fatty acid oxidation disorders (FAODs) are caused by defects in β-oxidation enzymes, including very long-chain acyl-CoA dehydrogenase (VLCAD), trifunctional protein (TFP), carnitine palmitoyltransferase-2 (CPT2), carnitine-acylcarnitine translocase (CACT) and others. During prolonged fasting, infection, or exercise, patients with FAODs present with hypoglycemia, rhabdomyolysis, cardiomyopathy, liver dysfunction, and occasionally sudden death. This article describes the diagnosis, newborn screening, and treatment of long-chain FAODs with a focus on VLCAD deficiency. VLCAD deficiency is generally classified into three phenotypes based on onset time, but the classification should be comprehensively determined based on genotype, residual enzyme activity, and clinical course, due to a lack of apparent genotype–phenotype correlation. With the expansion of newborn screening for FAODs, several issues have arisen, such as missed detection, overdiagnosis (including detection of benign/asymptomatic type), and poor prognosis of the neonatal-onset form. Meanwhile, dietary management and restriction of exercise have been unnecessary for patients with the benign/asymptomatic type of VLCAD deficiency with a high fatty acid oxidation flux score. Although L-carnitine therapy for VLCAD/TFP deficiency has been controversial, supplementation with L-carnitine may be accepted for CPT2/CACT and multiple acyl-CoA dehydrogenase deficiencies. Recently, a double-blind, randomized controlled trial of triheptanoin (seven-carbon fatty acid triglyceride) versus trioctanoin (regular medium-chain triglyceride) was conducted and demonstrated improvement of cardiac functions on triheptanoin. Additionally, although the clinical efficacy of bezafibrate remains controversial, a recent open-label clinical trial showed efficacy of this drug in improving quality of life. These drugs may be promising for the treatment of FAODs, though further studies are required.
Similar content being viewed by others
Introduction
Mitochondrial fatty acid oxidation disorders (FAODs) are a group of inherited metabolic diseases caused by defects in β-oxidation enzymes [1]. The β-oxidation enzymes involved include approximately 20 enzymes, such as very long-chain, medium-chain, and short-chain acyl-CoA dehydrogenases (VLCAD, MCAD, and SCAD, respectively), trifunctional protein (TFP, which consists of long-chain enoyl-CoA hydratase (LCEH), long-chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD), and long-chain 3-ketoacyl-CoA thiolase (LCKT)), carnitine palmitoyltransferase I and II (CPT1 and CPT2), carnitine-acylcarnitine translocase (CACT) and the carnitine transporter (Fig. 1). Electron transfer protein (ETF) and ETF dehydrogenase (ETFDH) are also involved. Fatty acids are mainly metabolized in the skeletal muscle, myocardium, and liver. If malfunction of these organs or tissues is triggered by prolonged fasting, vomiting, diarrhea, infectious illnesses, or heavy physical exercise, patients with FAODs often present with hypoglycemia, rhabdomyolysis, cardiomyopathy, arrhythmia, or liver dysfunction and occasionally with encephalopathy or unexpected sudden death [2].

Metabolic pathway of mitochondrial fatty acid oxidation. LCFAT long-chain fatty acid transporter, OCTN2 plasma membrane sodium-dependent carnitine transporter, AS acyl-CoA synthetase, CPT carnitine palmitoyltransferase, CACT carnitine-acylcarnitine translocase, VLCAD very long-chain acyl-CoA dehydrogenase, MCAD medium-chain acyl-CoA dehydrogenase, SCAD short-chain acyl-CoA dehydrogenase, TFP trifunctional protein, EH enoyl-CoA hydratase, HAD 3-hydroxyacyl-CoA dehydrogenase, KAT 3-ketoacyl-CoA thiolase, ETF electron transfer protein, ETFDH electron transfer protein dehydrogenase, e− electron, TCA cycle tricarboxylic acid cycle
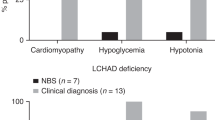
Among FAODs, deficiencies of long-chain fatty (LC) acids, such as VLCAD, TFP/LCHAD, CPT1/2, and CACT deficiencies, are referred to as LC-FAODs. The symptoms of each LC-FAOD are similar, and LC-FAODs are generally classified into three phenotypes, as shown in Table 1: the severe form, intermediate form, and myopathic form, all of which exhibit autosomal recessive inheritance. Moreover, a pre-symptomatic (or asymptomatic) form, identified by expanded newborn screening using acylcarnitine (AC) profiles by tandem mass spectrometry (ENBS) in a non-symptomatic condition, has been added. Herein, we describe recent clinical topics related to LC-FAODs, focusing mainly on VLCAD deficiency.
VLCAD deficiency
VLCAD deficiency (OMIM 201475, EC 1.3.99.13) is the most typical and well-known LC-FAOD. VLCAD is an enzyme that is located in the mitochondrial inner membrane (Fig. 1) and catalyzes the dehydrogenation of long-chain acyl-CoA esters of 12 to 18 carbons, which is the first step of β-oxidation [3,4,5,6]. The VLCAD enzyme is encoded by ACADVL, which comprises 20 exons spanning approximately 5.4 kb at 17p13.1 and forms as a homodimer of 70 kDa polypeptides [7, 8]. VLCAD deficiency shows an autosomal recessive inheritance pattern. Its prevalence differs among ethnic groups and is approximately 1:93,000 births in Japan [9], while it is estimated at 1:31,500 to 1:125,000 births in the Caucasian population [10,11,12]. More than 800 cases have been reported (clir-R4S.org consortium data) [13].
VLCAD deficiency is clinically classified into the following three groups, similar to other FAODs [14]. The severe early-onset form is characterized by hypertrophic or dilated cardiomyopathy, pericardial effusion, arrhythmia, hypotonia, hepatomegaly, or severe hypoketotic hypoglycemia in early infancy and is often fatal with cardiomyopathy and arrhythmias such as ventricular tachycardia, ventricular fibrillation, and atrioventricular block [15]. This form is also referred to as a cardiac and multiorgan failure VLCAD deficiency. However, such cardiac symptoms might be reversible with early intensive care and a medium-chain triglyceride (MCT) oil-based diet [16,17,18]. Furthermore, extracorporeal membrane oxygenation (ECMO) may be effective for cardiac symptoms, although there is no consensus about the efficacy of this treatment because of the few reports of ECMO in patients with the neonatal-onset form of VLCAD deficiency [19, 20]. However, one case report demonstrated that ECMO was effective for a neonate with MCAD deficiency who experienced cardiac arrest due to ventricular tachycardia and fibrillation [21], leading to the conclusion that there should be no hesitation in applying ECMO in patients with cardiomyopathy due to inherited metabolic diseases.
The milder/intermediate form of VLCAD deficiency, which is known as the hepatic or hypoketotic-hypoglycemic type, is typically associated with hypoketotic hypoglycemia and hepatomegaly induced by preceding infections or long fasting during early childhood. This form is characterized by a delayed onset, lower mortality, and scarce or absent cardiomyopathy compared with the severe form, which resembles MCAD deficiency [22]. However, long fasting or poor feeding occasionally induces acute symptoms during the newborn period, even in patients with the milder form [16]. Additionally, because encephalopathy or sudden infant death is sporadically reported even for the milder form [23,24,25,26], this form is not always “clinically milder”.
The later-onset type (myopathic form) presents mainly with episodic symptoms, consisting of skeletal muscle symptoms such as myalgia, muscle cramps/weakness, exercise intolerance, and/or rhabdomyolysis. These symptoms often appear in adolescence or adulthood during physical exercise or illness [27]. However, cardiomyopathy and respiratory failure can also be found in the later-onset form under special conditions [28, 29], and the p.V283A (sometimes referred to as p.V243A) mutation, known as a genotype of the later-onset type, may cause hypoglycemia [30, 31], suggesting that even the later-onset form can cause symptoms other than myopathy. Although it was previously reported that the neonatal-onset form was the most common type in VLCAD deficiency [14], the myopathic (or asymptomatic) form is likely the most common type according to recent ENBS data [16, 32, 33]. Indeed, among 29 Japanese patients with symptomatic VLCAD deficiency who were diagnosed in our laboratory (Shimane University), 23 patients had the myopathic from (data not published). In patients with this form, baseline creatine kinase (CK) levels generally return to normal after treatment, despite markedly elevated CK levels during the acute phase (occasionally > 100,000 IU/L);[34] however, some patients exhibit chronically high CK levels [35]. Moreover, some patients who are initially diagnosed with the neonatal-onset or milder form present only myopathic symptoms at a later age [33]. In short, the symptoms of VLCAD deficiency tend to improve with age regardless of the clinical form.
Overview of other LC-FAODs
TFP/LCHAD deficiency
Complete TFP deficiency involves defects in all three enzymes associated with the final three steps of LC-fatty acid β-oxidation (LCEH, LCHAD, and LCKT). Isolated LCHAD deficiency is due to a defect exclusively in LCHAD [36], while the other two enzymes, LCEH and LCKT are preserved. TFP deficiency and LCHAD deficiency show similar phenotypes and are usually grouped as TFP/LCHAD deficiency. However, it was recently reported that hypoparathyroidism is a common complication in Japanese patients with TFP deficiency but is not associated with LCHAD deficiency [37]. Moreover, LCHAD deficiency has never been detected in Japanese individuals, while the prevalence of LCHAD deficiency is relatively high in the Caucasian population, in which the common HADHA gene mutation, c.1528 G > C, is found [38]. The prognosis of TFP/LCHAD deficiency is likely more severe compared with that of VLCAD deficiency [33]. Neuropathy and retinopathy are specific for TFP/LCHAD deficiency and irreversible.
CPT2/CACT deficiency
CPT2 and CACT deficiencies present similar AC profiles and clinical symptoms, although CACT deficiency is collectively more severe compared with CPT2 deficiency. The myopathic form is frequently found in Caucasian patients with the common p.S113L mutation in the CPT2 gene [39]. We previously reported that CPT2 deficiency is an inherited metabolic disease that sometimes causes unexpected sudden death in Japan [40], suggesting that CPT2 deficiency should be carefully managed with attention to sudden infant death. In Japan, CPT2 deficiency was included in the core targets of ENBS since 2018. Although CACT deficiency is also a detectable disease associated with CPT2 deficiency in ENBS, it is very rare and has never been detected in ENBS of Japanese individuals so far [9]. There are a few case reports in Japan, while at least 30 patients with CACT deficiency have been described worlwide [41].
CPT1 deficiency
CPT1 deficiency is caused by a defect in CPT1A, which is locally expressed in the liver, kidney, lymphocytes, and fibroblasts. Therefore, muscle symptoms are not observed, unlike other LC-FAODs. The clinical phenotypes of CPT1 deficiency are rather similar to those of MCAD deficiency. The main symptoms include hypoketotic hypoglycemia, hyperammonemia, and fatty liver, but hepatic encephalopathy and sudden death have also been reported [42].
Multiple acyl-CoA dehydrogenase deficiency (MADD)
MADD is caused by a congenital defect in ETF or ETFDH, resulting in disturbance of the short- to long-chain acyl-CoA dehydrogenases and other mitochondrial dehydrogenases, such as glutaryl-CoA, isovaleryl-CoA, and sarcosine dehydrogenases, that are functionally regulated by ETF/ETFDH. MADD is also known as glutaric acidemia type II and is a unique FAOD that is categorized as a type of organic acidemia. Unlike other LC-FAODs, MADD is classically classified into distinct three phenotypes based on onset time and severity: neonatal onset with anomalies (type 1), neonatal onset without anomalies (type2), and mild and/or later onset (type 3) [43]. However, we previously proposed that MADD should be also classified into three forms (neonatal-onset, intermediate, and late-onset forms), similar to LC-FAODs [44], because the main symptoms of MADD are similar to those of LC-FAODs. The majority of patients with the late-onset type, particularly those with a defect in ETFDH, respond well to riboflavin administration [43].
Recently, other metabolic disorders involving riboflavin have been reported, such as riboflavin transporter deficiency, flavin adenine dinucleotide (FAD) transporter deficiency, or FAD synthase deficiency [45,46,47,48], which show similar biochemical and clinical findings to MADD. These diseases respond to riboflavin therapy. Therefore, when MADD is biochemically suspected, riboflavin should be administered first.
Diagnosis
Acylcarnitine analysis using tandem mass spectrometry
When the above symptoms suggestive of an FAOD, such as hypoketotic hypoglycemia, liver dysfunction, and rhabdomyolysis, are present, AC analysis is the first step in diagnosis. AC analysis reveals a specific profile for each disease. For example, VLCAD deficiency is suspected by an increase in tetradecenoylcarnitine (C14:1) and in the C14:1/C2, C14:1/C16, and C14:1/C12:1 ratios [49]. Although dried blood spots are usually used, serum/plasma AC analysis is more sensitive for the detection of LC-FAODs except for CPT1 deficiency [50,51,52]. However, the diagnosis should not be determined based on only clinical features and the blood AC profile. We identified a patient with asymptomatic VLCAD deficiency whose serum C14:1 levels were normal in a stable condition (not published). Additionally, AC analysis cannot distinguish CPT2 deficiency from CACT deficiency. Therefore, the following analysis is essential to confirm a definitive diagnosis.
Genetic analysis
Genetic analysis is most useful for diagnosing FAODs because it is relatively quick, precise, and convenient. Recently, a multigene panel evaluated through next-generation sequencing has also been applied for genetic diagnosis [53]. However, while genetic analysis is easy to perform, the interpretation of results, particularly with regard to novel mutation detection, is difficult. As a general rule, missense mutations cause milder phenotypes, and splice variants and nonsense mutations that are almost truncating result in severe phenotypes. However, LC-FAODs are actually heterogeneous diseases, and the phenotype-genotype correlation is poor, whereas it had been formerly believed that there was a clear phenotype-genotype correlation associated with VLCAD deficiency [14]. In fact, severe phenotypes may be presented even in cases involving missense mutations [54, 55]. Moreover, it has been reported that the onset time and symptoms differ among siblings [56, 57], indicating that it is difficult to accurately predict the clinical course and outcome by genetic information. Meanwhile, if only a single pathogenic variant is identified, heterozygous carriers cannot be distinguished from affected patients with non-detectable mutations. In cases where a definite diagnosis cannot be made by genetic analysis, functional assessment of β-oxidation and other biochemical tests, such as an enzyme activity assay, fatty acid oxidation (FAO) flux, in vitro probe (IVP) assay, and immunoblotting, may be helpful.
Enzyme assay, FAO flux, IVP assay, and immunoblotting
The methods of measuring residual β-oxidation enzyme, such as MCAD or VLCAD, activity using fibroblasts or lymphocytes were previously reported [58, 59]. Because the enzyme assay using lymphocytes is easy and quick, the combination of genetic and enzyme analyses is recommended for diagnosis of VLCAD, MCAD, and CPT2 deficiencies in Japan. Meanwhile, the activity of some enzymes, such as ETF, CACT, and carnitine transporter, is technically difficult to measure. Moreover, if residual enzyme activity is not particularly low in a case with novel mutations, confirming a definitive diagnosis will not be reached by the combination of genetic analysis and enzyme assay. For such cases, further specialized biochemical examinations may be necessary.
FAO flux is determined by measuring the production of radiolabeled H2O from [9,10-3H(N)]-oleic (or palmitic) acid using fibroblasts as a whole β-oxidation capacity [60]. While the FAO flux cannot be used to identify which enzyme is defective, it can be used to determine whether β-oxidation disorder is present or absent. Recently, it was reported that FAO flux is more strongly correlated with severity than VLCAD enzyme activity [54]. FAO flux may be a useful tool to predict the pathogenicity of novel mutations in ACADVL.
The IVP assay can indirectly estimate the β-oxidation capacity and defective sites of FAO, using fibroblasts cultured with unlabeled palmitate (carbon number is 16) and MS/MS for AC profiling [61]. In each FAOD, specific AC profiles can be observed. Elevation of LC-ACs (C10 to C16) is observed in cases of VLCAD deficiency, while C16 is elevated only in cases of CPT2/CACT deficiency (Fig. 2). Elevation in C16-OH, but a small peak, is characteristic of TFP/LCHAD deficiency in the IVP assay. Additionally, the IVP assay can distinguish the severity of some FAODs [62]. Whereas short- to long-chain ACs are elevated in milder form of MADD, only C16 accumulates in severe form of MADD, indicating that palmitate added as a substrate cannot be metabolized at all due to null mutations. In VLCAD deficiency, the peak of C16 tends to rise if the severity is significant, but other LC-ACs also rise even in severe forms. This result indicates that other acyl-CoA dehydrogenases, such as MCAD, compensate for the metabolism of long-chain fatty acids. Meanwhile, the IVP assay may not be sensitive enough to detect the myopathic form because the profile of the myopathic form is often scanty and similar to that of healthy controls [44]. However, the IVP assay is preferentially employed in our laboratory because of its usefulness not only for the diagnosis of FAODs but also for investigation of the effectiveness of several drugs and factors [63, 64].

Comparison of in vitro probe assays between individual phenotypes of each type of fatty acid oxidation disorder. The Y-axis represents the amount of acylcarnitines expressed in nmol/mg of protein. M, myopathic form; I, intermediate form; S severe form, VLCADD very long-chain acyl-CoA dehydrogenase deficiency, MADD multiple acyl-CoA dehydrogenase deficiency, TFPD trifunctional protein deficiency, CACTD carnitine-acylcarnitine transporter deficiency; Control: mean values from 4 healthy volunteers
Immunoblotting can be used for the diagnosis of FAODs when enzyme activity cannot be analyzed or such analyses are challenging, although specific antibodies are required. Immunoblotting reveals no band for either the TFP-α or TFP-β subunit in cases of TFP deficiency, despite a defect in either HADHA or HADHB [37]. Similarly, in MADD due to a defect in either ETFA or ETFB, both ETFA and ETFB bands are not present [44]. Therefore, genetic testing is also required for the definite diagnosis of such diseases. Although the pathogenicity of mutations can be partially predicted by in silico analysis [65], comprehensive analyses, such as the FAO flus, IVP assay and immunoblotting, are recommended to determine whether the mutation is truly pathogenic.
Problems in ENBS for VLCAD deficiency
ENBS for FAODs using tandem mass spectrometry has been implemented worldwide since the 1990s [12, 66], while nationwide ENBS has been implemented in Japan since 2014 [9]. Although the target diseases of ENBS have been a subject of debate [67], VLCAD deficiency is consistently recommended as a target of ENBS;[16] however, several problems remain to be solved.
Misdiagnosis and overdiagnosis
The first problem is the degree of accuracy. Affected patients are occasionally missed [10, 68, 69]. Physicians should not forget that AC profiles can be influenced by conditions such as time of sampling. To increase accuracy, various markers and the cutoffs are considered worldwide [70]. For example, elevated C14:1, C14:2, C14, C12, C14:1/C2, C14:1/C12:1, and C14:1/C16 ratios and combinations of these markers have been considered useful for the diagnosis of VLCAD deficiency [49]. Meanwhile, healthy individuals and heterozygous carriers can be sometimes detected as false-positives in ENBS [31, 71, 72]. In Japan, if the first ENBS using DBS performed on 4 to 6 days after birth indicates positive results for LC-FAODs, re-evaluation of serum AC is recommended to avoid missed detection because C14:1 levels in DBS gradually decrease after birth. Accordingly, a number of heterozygous carriers are detected because the sensitivity of serum AC analysis is too high. Therefore, when serum AC analysis shows positive results, enzyme activity assays and/or genetic testing should be immediately considered for definite diagnosis.
Moreover, “asymptomatic/benign VLCAD deficiency” detected by ENBS is problematic. The prevalence of VLCAD deficiency diagnosed by ENBS is significantly higher than that of clinically diagnosed VLCAD deficiency [12]. This finding suggests that a number of patients with the asymptomatic form are detected [67]. In many countries and regions, most patients with VLCAD deficiency detected by ENBS remain asymptomatic during follow-up for at least several years [16, 33, 49]. However, some initially asymptomatic children manifest myopathic symptoms after extensive physical exercise at a later age, despite early treatment [22]. This result suggests that early intervention may not be effective for preventing the onset of myopathy; i.e., it may be meaningless to detect myopathic VLCAD deficiency by ENBS. Additionally, Ryder B, et al. reported that a mutation (p.T409M) that is commonly found in Maori or Pacific populations in New Zealand is a benign variant of VLCAD deficiency, despite the presence of elevated C14:1 levels in ENBS [73]. This study indicates that mutations, which have never been identified in the database of clinically diagnosed cases but are often identified by ENBS, are likely benign variants. Although the problem of “overdiagnosis of asymptomatic/benign disease” may be excused at present, over treatment and intervention should be avoided for such patients.
One additional problem could occur in familial screening. When VLCAD deficiency is detected in a baby through ENBS, older children in the family should undergo sibling screening, regardless of the presence or absence of symptoms. If the older children also have asymptomatic/benign VLCAD deficiency, it will be very difficult to determine what management strategy should be applied to the siblings who are strongly suspected of remaining asymptomatic without treatment.
Poor outcomes of the neonatal-onset form
The second problem is the difficulty of rescuing patients with the neonatal-onset form. Twenty-three of a group of 37 Saudi Arabian patients with VLCAD deficiency including 31 patients with a homozygous nonsense mutation (p.Ser22X) died before the age of 2 years, despite early detection and early intervention with MCT supplementation [74]. One infantile case involving a patient with the neonatal-onset form who died suddenly due to respiratory syncytial virus infection, in spite of early detection, has also been reported [75]. These findings may indicate that ENBS contributes little to improving the survivability of cases with the neonatal-onset form. New approaches or management strategies other than MCT are expected to be developed in the future.
In conclusion, the two above problems are applicable not only to VLCAD deficiency but also to a number of inherited metabolic diseases detected by ENBS. However, ENBS significantly prevents infantile death due to inherited metabolic diseases. Moreover, Landau YE, et al. reported that patients who have been clinically diagnosed during the ENBS era are identified at younger ages because of the spread of information about metabolic diseases [67]. Briefly, ENBS is useful not only for diagnosis but also for education.
Current topics concerning treatments for LC-FAODs
There is no permanent treatment, such as gene therapy, for LC-FAODs. The most important treatment strategy is avoidance of triggers (e.g., fever, diarrhea, vomiting, hard exercise, prolonged fasting, and/or overloading of long-chain fatty acids) and the use of intravenous glucose as an energy source during the acute phase [76]. Intravenous glucose infusions on “sick days”, such as during infection and loss of appetite, are useful for preventing severe metabolic decompensation.
Dietary management
The avoidance of prolonged fasting, restriction of LC-fatty acids, and supplementation with MCT are generally recommended as dietary management strategies. The acceptable maximum fasting periods according to Japanese guideline are modified based on European recommendations [77]. The period should be shortened during an illness or in severe symptomatic patients. However, restriction of LC-fatty acids and supplementation with MCT are presumably unnecessary in patients with asymptomatic VLCAD deficiency. Moreover, even avoidance of prolonged fasting may not be needed for VLCAD-deficient patients with LC-FAO flux score greater than 90% of normal, who are strongly predicted to remain asymptomatic [78]. In contrast, all patients with FAODs excluding such asymptomatic VLCAD deficiency should comply with the avoidance of prolonged fasting, even if they are asymptomatic at diagnosis because other LC-FAODs are generally more severe than VLCAD deficiency.
Restriction of exercise
Extensive or sustained exercise (e.g., military training or climbing a mountain) may trigger metabolic decompensation in patients with LC-FAODs. Additionally, some patients with an initially asymptomatic type of VLCAD deficiency may develop muscle symptoms during exercise in later life, despite treatment [27, 79]. However, restriction of exercise is not always necessary for patients with LC-FAODs. Because pre-exercise supplementation with MCT improves exercise tolerance among patients with LC-FAODs [80], participating sports and routine physical activity should not be avoided for patients who can implement pre-exercise supplementation with MCT. In contrast, oral glucose supplementation immediately before exercise might worsen the exercise capacity, most likely due to the sympathoadrenal response, which increases the heart rate and blocks gluconeogenesis [81]. Nevertheless, glucose supplementation, rest, and rehydration are important during exercise. Exercise restriction should be determined based on an individual’s exercise tolerance level.
Supplementation with L-carnitine
L-carnitine supplementation for FAODs is considered to maintain the serum-free carnitine concentration and eliminate toxic ACs [82]. However, the use of L-carnitine supplementation is controversial at present. For example, it is not recommended for either LC-FAODs or MCAD deficiency because the beneficial effect of L-carnitine supplementation is not proven [76]. In particular, L-carnitine supplementation should be avoided at times of severe metabolic dysfunction in patients with VLCAD and TFP/LCHAD deficiencies because of a risk of arrhythmia provoked by the accumulation of LC-ACs [77]. We previously described two siblings with VLCAD deficiency suffering from recurrent rhabdomyolysis only during L-carnitine supplementation [57]. This case report suggested that L-carnitine supplementation may cause the pathological conditions of patients with VLCAD deficiency to deteriorate. By contrast, it has been reported that L-carnitine supplementation does not increase LC-AC levels in patients with LC-FAODs on a triheptanoin diet, while it facilitates the export of excessive toxic LC-acyl-CoA intermediates as ACs and preserves the levels of free CoA [83].
Regarding how L-carnitine supplementation should be viewed in the context of MADD or CPT2/CACT deficiency, there are few recommendations. Because these diseases often result in severe secondary carnitine deficiency, a certain level of L-carnitine supplementation may be necessary. Indeed, for MADD, L-carnitine supplementation is not always discouraged [84, 85]. Additionally, we found that the frequency of rhabdomyolysis decreased after high-dose L-carnitine supplementation in a patient with CPT2 deficiency, although other treatments, such as dietary management, MCT, and bezafibrate, might have had synergistic effects [86]. However, it should be considered that L-carnitine supplementation could result in the accumulation of LC-ACs even in patients with these diseases. Hence, further studies on the application of L-carnitine therapy for the above diseases are required.
Triheptanoin
Triheptanoin, a seven-carbon fatty acid triglyceride (C7), is a promising drug for LC-FAODs. Anaplerotic metabolites of C7 are hypothesized to have the potential to replace deficient Krebs cycle intermediates through conversion to succinyl-CoA, resulting in net glucose production as a novel energy source [87]. Recently, a double-blind randomized controlled trial of the use of C7 versus trioctanoin (regular MCT) in LC-FAODs was conducted in the United States [88]. This trial concluded that C7 significantly improved the left ventricle (LV) ejection fraction and reduced the LV mass at rest and the heart rate during exercise compared to regular MCT. In a retrospective study, the same research group revealed that C7 decreased the duration and rate of hospitalization and the frequency of hypoglycemia [89] and improved the endurance and tolerance of exercise and quality of life (QOL) in an open-label trial [87]. The adverse effects of C7 consist almost exclusively of gastrointestinal symptoms, such as diarrhea, nausea, and abdominal pain, although the frequency of adverse events is similar to that of regular MCT. In contrast, in VLCAD knockout mice, long-term application of dietary C7 was found to have no positive effect and failed to prevent the development of systolic dysfunction in the cardiac phenotype [90]. Although CACT and CPT-1 deficiencies were excluded in the above studies, and the efficacy of C7 for the neonatal-onset type of LC-FAODs is unknown in humans, C7 may improve several clinical manifestations compared with regular MCT in all patients with LC-FAODs.
Bezafibrate
Bezafibrate [2-(p-(2-(p-chlorobenzamido)ethyl)-phenoxy)-2-methyl propionic acid] is a peroxisome proliferator-activated receptor agonist that decreases human serum lipid levels. By enhancing the transcription of several β-oxidation enzymes in vitro, bezafibrate has been reported as a promising drug for FAODs [91,92,93]. We previously reported the effect of bezafibrate for several FAODs in in vitro studies and case reports [41, 64, 86, 94, 95]. However, its in vivo efficacy remains controversial, although the in vitro efficacy of bezafibrate has been reported from numerous studies.
As shown in Table 2, three clinical trials of bezafibrate have been conducted to date. A French group (Bonnefont JP, et al.) first reported the efficacy and long-term safety of bezafibrate in patients with the myopathic form of CPT2 deficiency [96, 97]. These clinical trials revealed that bezafibrate increased the level of oxidation of palmitoyl L-carnitine in isolated muscle mitochondria, the expression of CPT2 messenger RNA and protein in the skeletal muscle, and subjective QOL scores based on the SF-36 questionnaire, while it decreased the number of rhabdomyolysis episodes and plasma CK levels. Recently, we also reported that bezafibrate significantly improved subjective QOL scores in a small population trial whose study design imitated those of the open-label French trials [98]. Although the primary end point (the frequency of myopathic attacks) and the other outcomes (e.g., levels of serum CK, ACs, and myalgia during myopathic attacks) were not changed in our trial, we concluded that these end points could not be evaluated due to several limitations, such as problems in the definition of myopathic attack. Only QOL scores using SF-36 were significantly improved, but this result may have been due to the placebo effect because of the open-label trial design. In contrast, a Danish group (Ørngreen MC, et al.) reported that bezafibrate did not improve clinical symptoms or FAO capacity during exercise in patients with CPT2 and VLCAD deficiencies [99]. Because this trial was designed as a double-blind, randomized, placebo-controlled, parallel, crossover study, meaning that its results constitute Class 1 evidence, the clinical efficacy of bezafibrate has been questioned. The French group had a heated debate about the end points and investigations applied in this trial with the Danish group [100, 101]. Because the participants in this trial experienced almost no rhabdomyolysis episodes before and after the administration of bezafibrate, we also doubt that such subjects are appropriate for evaluating its clinical efficacy. Based on the results of our trial, more than half of the participants subjectively experienced the efficacy of bezafibrate, even if the frequency of myopathic attacks was not reduced. However, these subjective feelings are not easy to quantify and estimate, particularly in open-label trials. Because it cannot be concluded that bezafibrate does not currently have any clinical efficacy, further double-blind, randomized, placebo-controlled clinical trials in which QOL is included as an endpoint are necessary to elucidate the efficacy of bezafibrate.
In addition, bezafibrate is considered to be ineffective for patients with the neonatal-onset form, such as subjects with null mutations even in in vitro. Therefore, it will be preferable to evaluate the efficacy of bezafibrate in vitro if its administration is initiated. For the prediction of in vitro efficacy, FAO flux is a better tool than the IVP assay. As shown in Fig. 3, the IVP assay revealed that the accumulation of C16 decreased in the presence of high-dose bezafibrate, apparently indicating that bezafibrate is effective even for the severe form of MADD. By contrast, FAO flux showed no efficacy in the severe form. Because this MADD patient with the severe form had a null mutation and bezafibrate was not effective in this individual, the result of the IVP assay for the severe form was considered an error. These findings suggest that overdose of bezafibrate might deteriorate FAO flux and production of ACs although the causes are unknown.

Comparison of the IVP assay and FAO flux in fibroblasts derived from patients with milder and severe forms of multiple acyl-CoA dehydrogenase deficiency. a Changes in acylcarnitine profiles in response to 100 or 800 µM bezafibrate in an in vitro probe (IVP) assay. b Results of fatty acid oxidation (FAO) flux assessment using the radio-isotope palmitic acid in the absence and presence of bezafibrate (100 and 800 µM). Bez0, absence of bezafibrate; Bez100, 100 µM dose of bezafibrate; Bez800, 800 µM dose of bezafibrate
Conclusion
With the expansion of ENBS for FAODs, an increasing number of patients with milder phenotypes, including asymptomatic form, have been identified. Because the outcomes of such patients cannot be accurately predicted, careful management without overtreatment is required. In the myopathic or asymptomatic form of VLCAD deficiency, dietary therapy and exercise restriction can be more moderate. While the efficacy of bezafibrate remains controversial, triheptanoin has shown therapeutic potential in patients with FAODs; however, the extent of the improvement of clinical symptoms or the reduction of the degree of management remains unknown. The prognosis of the neonatal-onset form remains poor, despite early diagnosis and intervention. The accumulation of evidence obtained from clinical and basic investigations will reveal strategies for addressing issues associated with the management of FAODs.
References
Vishwanath VA. Fatty acid beta-oxidation disorders: a brief review. Ann Neurosci. 2016;23:51–55.
Shekhawat PS, Matern D, Strauss AW. Fetal fatty acid oxidation disorders, their effect on maternal health and neonatal outcome: impact of expanded newborn screening on their diagnosis and management. Pediatr Res. 2005;57:78r–86.
Izai K, Uchida Y, Orii T, Yamamoto S, Hashimoto T. Novel fatty acid beta-oxidation enzymes in rat liver mitochondria. I. Purification and properties of very-long-chain acyl-coenzyme A dehydrogenase. J Biol Chem. 1992;267:1027–33.
Yamaguchi S, Indo Y, Coates PM, Hashimoto T, Tanaka K. Identification of very-long-chain acyl-CoA dehydrogenase deficiency in three patients previously diagnosed with long-chain acyl-CoA dehydrogenase deficiency. Pediatr Res. 1993;34:111–3.
Aoyama T, Uchida Y, Kelley RI, Marble M, Hofman K, Tonsgard JH, et al. A novel disease with deficiency of mitochondrial very-long-chain acyl-CoA dehydrogenase. Biochem Biophys Res Commun. 1993;191:1369–72.
Souri M, Aoyama T, Yamaguchi S, Hashimoto T. Relationship between structure and substrate-chain-length specificity of mitochondrial very-long-chain acyl-coenzyme A dehydrogenase. Eur J Biochem. 1998;257:592–8.
McAndrew RP, Wang Y, Mohsen AW, He M, Vockley J, Kim JJ. Structural basis for substrate fatty acyl chain specificity: crystal structure of human very-long-chain acyl-CoA dehydrogenase. J Biol Chem. 2008;283:9435–43.
Souri M, Aoyama T, Hoganson G, Hashimoto T. Very-long-chain acyl-CoA dehydrogenase subunit assembles to the dimer form on mitochondrial inner membrane. FEBS Lett. 1998;426:187–90.
Shibata N, Hasegawa Y, Yamada K, Kobayashi H, Purevsuren J, Yang Y, et al. Diversity in the incidence and spectrum of organic acidemias, fatty acid oxidation disorders, and amino acid disorders in Asian countries: Selective screening vs. expanded newborn screening. Mol Genet Metab Rep. 2018;16:5–10.
Boneh A, Andresen BS, Gregersen N, Ibrahim M, Tzanakos N, Peters H, et al. VLCAD deficiency: pitfalls in newborn screening and confirmation of diagnosis by mutation analysis. Mol Genet Metab. 2006;88:166–70.
Spiekerkoetter U, Sun B, Zytkovicz T, Wanders R, Strauss AW, Wendel U. MS/MS-based newborn and family screening detects asymptomatic patients with very-long-chain acyl-CoA dehydrogenase deficiency. J Pediatr. 2003;143:335–42.
Wilcken B, Wiley V, Hammond J, Carpenter K. Screening newborns for inborn errors of metabolism by tandem mass spectrometry. N Engl J Med. 2003;348:2304–12.
Leslie ND, Valencia CA, Strauss AW, Zhang K. Very long-chain acyl-Coenzyme A dehydrogenase deficiency. 2009 May 28 [updated 2018 Jan 4]. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2018. Available from http://www.ncbi.nlm.nih.gov/books/NBK6816/ PubMed PMID: 20301763.
Andresen BS, Olpin S, Poorthuis BJ, Scholte HR, Vianey-Saban C, Wanders R, et al. Clear correlation of genotype with disease phenotype in very-long-chain acyl-CoA dehydrogenase deficiency. Am J Hum Genet. 1999;64:479–94.
Bonnet D, Martin D, Pascale De L, Villain E, Jouvet P, Rabier D, et al. Arrhythmias and conduction defects as presenting symptoms of fatty acid oxidation disorders in children. Circulation. 1999;100:2248–53.
Pena LD, van Calcar SC, Hansen J, Edick MJ, Walsh Vockley C, Leslie N, et al. Outcomes and genotype-phenotype correlations in 52 individuals with VLCAD deficiency diagnosed by NBS and enrolled in the IBEM-IS database. Mol Genet Metab. 2016;118:272–81.
Vockley J, Charrow J, Ganesh J, Eswara M, Diaz GA, McCracken E, et al. Triheptanoin treatment in patients with pediatric cardiomyopathy associated with long chain-fatty acid oxidation disorders. Mol Genet Metab. 2016;119:223–31.
Sharef SW, Al-Senaidi K, Joshi SN. Successful treatment of cardiomyopathy due to very long-chain Acyl-CoA dehydrogenase deficiency: first case report from Oman with literature review. Oman Med J. 2013;28:354–6.
Katz S, Landau Y, Pode-Shakked B, Pessach IM, Rubinshtein M, Anikster Y, et al. Cardiac failure in very long chain acyl-CoA dehydrogenase deficiency requiring extracorporeal membrane oxygenation (ECMO) treatment: A case report and review of the literature. Mol Genet Metab Rep. 2017;10:5–7.
Mathur A, Sims HF, Gopalakrishnan D, Gibson B, Rinaldo P, Vockley J, et al. Molecular heterogeneity in very-long-chain acyl-CoA dehydrogenase deficiency causing pediatric cardiomyopathy and sudden death. Circulation. 1999;99:1337–43.
Kumar G, Mattke AC, Bowling F, McWhinney A, Alphonso N, Karl TR. Resuscitation of a neonate with medium chain acyl-coenzyme a dehydrogenase deficiency using extracorporeal life support. World J Pediatr Congenit Heart Surg. 2014;5:118–20.
Spiekerkoetter U. Mitochondrial fatty acid oxidation disorders: clinical presentation of long-chain fatty acid oxidation defects before and after newborn screening. J Inherit Metab Dis. 2010;33:527–32.
Ohashi Y, Hasegawa Y, Murayama K, Ogawa M, Hasegawa T, Kawai M, et al. A new diagnostic test for VLCAD deficiency using immunohistochemistry. Neurology. 2004;62:2209–13.
Takahashi Y, Sano R, Nakajima T, Kominato Y, Kubo R, Takahashi K, et al. Combination of postmortem mass spectrometry imaging and genetic analysis reveals very long-chain acyl-CoA dehydrogenase deficiency in a case of infant death with liver steatosis. Forensic Sci Int. 2014;244:e34–37.
Scalais E, Bottu J, Wanders RJ, Ferdinandusse S, Waterham HR, De Meirleir L. Familial very long chain acyl-CoA dehydrogenase deficiency as a cause of neonatal sudden infant death: improved survival by prompt diagnosis. Am J Med Genet A. 2015;167a:211–4.
Coughlin CR 2nd, Ficicioglu C. Genotype-phenotype correlations: sudden death in an infant with very-long-chain acyl-CoA dehydrogenase deficiency. J Inherit Metab Dis. 2010;33(Suppl 3):S129–131.
Laforet P, Acquaviva-Bourdain C, Rigal O, Brivet M, Penisson-Besnier I, Chabrol B, et al. Diagnostic assessment and long-term follow-up of 13 patients with very long-chain acyl-coenzyme A dehydrogenase (VLCAD) deficiency. Neuromuscul Disord. 2009;19:324–9.
Ficicioglu C, Hussa C. Very long-chain acyl-CoA dehydrogenase deficiency: the effects of accidental fat loading in a patient detected through newborn screening. J Inherit Metab Dis. 2009;32(Suppl 1):S187–190.
Tong MK, Lam CS, Mak TW, Fu MY, Ng SH, Wanders RJ, et al. Very long-chain acyl-CoA dehydrogenase deficiency presenting as acute hypercapnic respiratory failure. Eur Respir J. 2006;28:447–50.
Liebig M, Schymik I, Mueller M, Wendel U, Mayatepek E, Ruiter J, et al. Neonatal screening for very long-chain acyl-coA dehydrogenase deficiency: enzymatic and molecular evaluation of neonates with elevated C14:1-carnitine levels. Pediatrics. 2006;118:1065–9.
Miller MJ, Burrage LC, Gibson JB, Strenk ME, Lose EJ, Bick DP, et al. Recurrent ACADVL molecular findings in individuals with a positive newborn screen for very long chain acyl-coA dehydrogenase (VLCAD) deficiency in the United States. Mol Genet Metab. 2015;116:139–45.
Kang E, Kim YM, Kang M, Heo SH, Kim GH, Choi IH, et al. Clinical and genetic characteristics of patients with fatty acid oxidation disorders identified by newborn screening. BMC Pediatr. 2018;18:103.
Spiekerkoetter U, Lindner M, Santer R, Grotzke M, Baumgartner MR, Boehles H, et al. Management and outcome in 75 individuals with long-chain fatty acid oxidation defects: results from a workshop. J Inherit Metab Dis. 2009;32:488–97.
Yamamoto F, Nakamagoe K, Yamada K, Ishii A, Furuta J, Yamaguchi S, et al. A case of very-long-chain acyl-coenzyme A dehydrogenase deficiency with novel compound heterozygous mutations. J Neurol Sci. 2016;368:165–7.
Shchelochkov O, Wong LJ, Shaibani A, Shinawi M. Atypical presentation of VLCAD deficiency associated with a novel ACADVL splicing mutation. Muscle Nerve. 2009;39:374–82.
Das AM, Illsinger S, Lucke T, Hartmann H, Ruiter JP, Steuerwald U, et al. Isolated mitochondrial long-chain ketoacyl-CoA thiolase deficiency resulting from mutations in the HADHB gene. Clin Chem. 2006;52:530–4.
Bo R, Yamada K, Kobayashi H, Jamiyan P, Hasegawa Y, Taketani T, et al. Clinical and molecular investigation of 14 Japanese patients with complete TFP deficiency: a comparison with Caucasian cases. J Hum Genet. 2017;62:809–14.
Boutron A, Acquaviva C, Vianey-Saban C, de Lonlay P, de Baulny HO, Guffon N, et al. Comprehensive cDNA study and quantitative analysis of mutant HADHA and HADHB transcripts in a French cohort of 52 patients with mitochondrial trifunctional protein deficiency. Mol Genet Metab. 2011;103:341–8.
Joshi PR, Deschauer M, Zierz S. Carnitine palmitoyltransferase II (CPT II) deficiency: genotype-phenotype analysis of 50 patients. J Neurol Sci. 2014;338:107–11.
Takahashi T, Yamada K, Kobayashi H, Hasegawa Y, Taketani T, Fukuda S, et al. Metabolic disease in 10 patients with sudden unexpected death in infancy or acute life-threatening events. Pediatr Int. 2015;57:348–53.
Vatanavicharn N, Yamada K, Aoyama Y, Fukao T, Densupsoontorn N, Jirapinyo P, et al. Carnitine-acylcarnitine translocase deficiency: Two neonatal cases with common splicing mutation and in vitro bezafibrate response. Brain Dev. 2015;37:698–703.
Collins SA, Sinclair G, McIntosh S, Bamforth F, Thompson R, Sobol I, et al. Carnitine palmitoyltransferase 1A (CPT1A) P479L prevalence in live newborns in Yukon, Northwest Territories, and Nunavut. Mol Genet Metab. 2010;101:200–4.
Grunert SC. Clinical and genetical heterogeneity of late-onset multiple acyl-coenzyme A dehydrogenase deficiency. Orphanet J Rare Dis. 2014;9:117.
Yamada K, Kobayashi H, Bo R, Takahashi T, Purevsuren J, Hasegawa Y, et al. Clinical, biochemical and molecular investigation of adult-onset glutaric acidemia type II: characteristics in comparison with pediatric cases. Brain Dev. 2016;38:293–301.
Olsen RKJ, Konarikova E, Giancaspero TA, Mosegaard S, Boczonadi V, Matakovic L, et al. Riboflavin-responsive and non-responsive mutations in FAD synthase cause multiple Acyl-CoA dehydrogenase and combined respiratory-chain deficiency. Am J Hum Genet. 2016;98:1130–45.
Auranen M, Paetau A, Piirila P, Pohju A, Salmi T, Lamminen A, et al. Patient with multiple acyl-CoA dehydrogenation deficiency disease and FLAD1 mutations benefits from riboflavin therapy. Neuromuscul Disord. 2017;27:581–4.
Schiff M, Veauville-Merllie A, Su CH, Tzagoloff A, Rak M, Ogier de Baulny H, et al. SLC25A32 mutations and riboflavin-responsive exercise intolerance. N Engl J Med. 2016;374:795–7.
Bosch AM, Abeling NG, Ijlst L, Knoester H, van der Pol WL, Stroomer AE, et al. Brown-Vialetto-Van Laere and Fazio Londe syndrome is associated with a riboflavin transporter defect mimicking mild MADD: a new inborn error of metabolism with potential treatment. J Inherit Metab Dis. 2011;34:159–64.
Merinero B, Alcaide P, Martin-Hernandez E, Morais A, Garcia-Silva MT, Quijada-Fraile P, et al. Four years’ experience in the diagnosis of very long-chain Acyl-CoA dehydrogenase deficiency in infants detected in three Spanish newborn screening centers. JIMD Rep. 2018;39:63–74.
Topcu Y, Bayram E, Karaoglu P, Yis U, Kurul SH. Importance of acylcarnitine profile analysis for disorders of lipid metabolism in adolescent patients with recurrent rhabdomyolysis: Report of two cases. Ann Indian Acad Neurol. 2014;17:437–40.
Al-Thihli K, Sinclair G, Sirrs S, Mezei M, Nelson J, Vallance H. Performance of serum and dried blood spot acylcarnitine profiles for detection of fatty acid beta-oxidation disorders in adult patients with rhabdomyolysis. J Inherit Metab Dis. 2014;37:207–13.
Vreken P, van Lint AE, Bootsma AH, Overmars H, Wanders RJ, van Gennip AH. Quantitative plasma acylcarnitine analysis using electrospray tandem mass spectrometry for the diagnosis of organic acidaemias and fatty acid oxidation defects. J Inherit Metab Dis. 1999;22:302–6.
Smon A, Repic Lampret B, Groselj U, Zerjav Tansek M, Kovac J, Perko D, et al. Next generation sequencing as a follow-up test in an expanded newborn screening programme. Clin Biochem. 2018;52:48–55.
Diekman EF, Ferdinandusse S, van der Pol L, Waterham HR, Ruiter JP, Ijlst L, et al. Fatty acid oxidation flux predicts the clinical severity of VLCAD deficiency. Genet Med. 2015;17:989–94.
Goetzman ES, Wang Y, He M, Mohsen AW, Ninness BK, Vockley J. Expression and characterization of mutations in human very long-chain acyl-CoA dehydrogenase using a prokaryotic system. Mol Genet Metab. 2007;91:138–47.
Straussberg R, Strauss AW. A novel mutation of late-onset very-long-chain acyl-CoA dehydrogenase deficiency. Pediatr Neurol. 2002;27:136–7.
Watanabe K, Yamada K, Sameshima K, Yamaguchi S. Two siblings with very long-chain acyl-CoA dehydrogenase (VLCAD) deficiency suffered from rhabdomyolysis after l-carnitine supplementation. Mol Genet Metab Rep. 2018;15:121–3.
Tajima G, Sakura N, Yofune H, Nishimura Y, Ono H, Hasegawa Y, et al. Enzymatic diagnosis of medium-chain acyl-CoA dehydrogenase deficiency by detecting 2-octenoyl-CoA production using high-performance liquid chromatography: a practical confirmatory test for tandem mass spectrometry newborn screening in Japan. J Chromatogr B Anal Technol Biomed Life Sci. 2005;823:122–30.
Tajima G, Sakura N, Shirao K, Okada S, Tsumura M, Nishimura Y, et al. Development of a new enzymatic diagnosis method for very-long-chain Acyl-CoA dehydrogenase deficiency by detecting 2-hexadecenoyl-CoA production and its application in tandem mass spectrometry-based selective screening and newborn screening in Japan. Pediatr Res. 2008;64:667–72.
Manning NJ, Olpin SE, Pollitt RJ, Webley J. A comparison of [9,10-3H]palmitic and [9,10-3H]myristic acids for the detection of defects of fatty acid oxidation in intact cultured fibroblasts. J Inherit Metab Dis. 1990;13:58–68.
Okun JG, Kolker S, Schulze A, Kohlmuller D, Olgemoller K, Lindner M, et al. A method for quantitative acylcarnitine profiling in human skin fibroblasts using unlabelled palmitic acid: diagnosis of fatty acid oxidation disorders and differentiation between biochemical phenotypes of MCAD deficiency. Biochim Biophys Acta. 2002;1584:91–98.
Endo M, Hasegawa Y, Fukuda S, Kobayashi H, Yotsumoto Y, Mushimoto Y, et al. In vitro probe acylcarnitine profiling assay using cultured fibroblasts and electrospray ionization tandem mass spectrometry predicts severity of patients with glutaric aciduria type 2. J Chromatogr B Anal Technol Biomed Life Sci. 2010;878:1673–6.
Li H, Fukuda S, Hasegawa Y, Purevsuren J, Kobayashi H, Mushimoto Y, et al. Heat stress deteriorates mitochondrial beta-oxidation of long-chain fatty acids in cultured fibroblasts with fatty acid beta-oxidation disorders. J Chromatogr B Anal Technol Biomed Life Sci. 2010;878:1669–72.
Yamada K, Kobayashi H, Bo R, Purevsuren J, Mushimoto Y, Takahashi T, et al. Efficacy of bezafibrate on fibroblasts of glutaric acidemia type II patients evaluated using an in vitro probe acylcarnitine assay. Brain Dev. 2017;39:48–57.
Catarzi S, Caciotti A, Thusberg J, Tonin R, Malvagia S, la Marca G, et al. Medium-chain acyl-CoA deficiency: outlines from newborn screening, in silico predictions, and molecular studies. Sci World J. 2013;2013:625824.
Wilcken B. Newborn screening: how are we travelling, and where should we be going? J Inherit Metab Dis. 2011;34:569–74.
Landau YE, Waisbren SE, Chan LM, Levy HL. Long-term outcome of expanded newborn screening at Boston children’s hospital: benefits and challenges in defining true disease. J Inherit Metab Dis. 2017;40:209–18.
Estrella J, Wilcken B, Carpenter K, Bhattacharya K, Tchan M, Wiley V. Expanded newborn screening in New South Wales: missed cases. J Inherit Metab Dis. 2014;37:881–7.
Ficicioglu C, Coughlin CR 2nd, Bennett MJ, Yudkoff M. Very long-chain acyl-CoA dehydrogenase deficiency in a patient with normal newborn screening by tandem mass spectrometry. J Pediatr. 2010;156:492–4.
McHugh D, Cameron CA, Abdenur JE, Abdulrahman M, Adair O, A Nuaimi SA, et al. Clinical validation of cutoff target ranges in newborn screening of metabolic disorders by tandem mass spectrometry: a worldwide collaborative project. Genet Med. 2011;13:230–54.
Shigematsu Y, Hirano S, Hata I, Tanaka Y, Sudo M, Tajima T, et al. Selective screening for fatty acid oxidation disorders by tandem mass spectrometry: difficulties in practical discrimination. J Chromatogr B Anal Technol Biomed Life Sci. 2003;792:63–72.
Schiff M, Mohsen AW, Karunanidhi A, McCracken E, Yeasted R, Vockley J. Molecular and cellular pathology of very-long-chain acyl-CoA dehydrogenase deficiency. Mol Genet Metab. 2013;109:21–27.
Ryder B, Knoll D, Love DR, Shepherd P, Love JM, Reed PW, et al. The natural history of elevated tetradecenoyl-L-carnitine detected by newborn screening in New Zealand: implications for very long chain acyl-CoA dehydrogenase deficiency screening and treatment. J Inherit Metab Dis. 2016;39:409–14.
aid A, Nashabat M, Alfadhel M, Alasmari A, Al Mutairi F, Alswaid A, et al. Clinical, biochemical, and molecular features in 37 Saudi Patients with very long chain Acyl CoA dehydrogenase deficiency. JIMD Rep. 2018;40:47–53.
Yamamoto A, Nakamura K, Matsumoto S, Iwai M, Shigematsu Y, Tajima G, et al. VLCAD deficiency in a patient who recovered from ventricular fibrillation, but died suddenly of a respiratory syncytial virus infection. Pediatr Int. 2013;55:775–8.
Spiekerkoetter U, Bastin J, Gillingham M, Morris A, Wijburg F, Wilcken B. Current issues regarding treatment of mitochondrial fatty acid oxidation disorders. J Inherit Metab Dis. 2010;33:555–61.
Spiekerkoetter U, Lindner M, Santer R, Grotzke M, Baumgartner MR, Boehles H, et al. Treatment recommendations in long-chain fatty acid oxidation defects: consensus from a workshop. J Inherit Metab Dis. 2009;32:498–505.
Bleeker, JC, Kok, IL, Ferdinandusse, S, de Vries, M, Derks, TGJ, Mulder, MF et al. Proposal for an individualized dietary strategy in patients with very long-chain acyl-CoA dehydrogenase deficiency. J. Inherit. Metab. Dis. (2018).
Spiekerkoetter U. Effects of a fat load and exercise on asymptomatic VLCAD deficiency. J Inherit Metab Dis. 2007;30:405.
Gillingham MB, Scott B, Elliott D, Harding CO. Metabolic control during exercise with and without medium-chain triglycerides (MCT) in children with long-chain 3-hydroxy acyl-CoA dehydrogenase (LCHAD) or trifunctional protein (TFP) deficiency. Mol Genet Metab. 2006;89:58–63.
Orngreen MC. Substrate kinetics in patients with disorders of skeletal muscle metabolism. Dan. Med. J. 2016;63.
Winter SC. Treatment of carnitine deficiency. J Inherit Metab Dis. 2003;26:171–80.
Roe CR, Brunengraber H. Anaplerotic treatment of long-chain fat oxidation disorders with triheptanoin: review of 15 years Experience. Mol Genet Metab. 2015;116:260–8.
Creanza A, Cotugno M, Mazzaccara C, Frisso G, Parenti G & Capaldo B. Successful pregnancy in a young woman with multiple Acyl-CoA dehydrogenase deficiency. JIMD Rep. 2018;39:1–6.
Prasad M, Hussain S. Glutaric aciduria type II presenting as myopathy and rhabdomyolysis in a teenager. J Child Neurol. 2015;30:96–99.
Shima A, Yasuno T, Yamada K, Yamaguchi M, Kohno R, Yamaguchi S, et al. First Japanese case of carnitine palmitoyltransferase II deficiency with the homozygous point mutation S113L. Intern Med. 2016;55:2659–61.
Vockley J, Burton B, Berry GT, Longo N, Phillips J, Sanchez-Valle A, et al. UX007 for the treatment of long chain-fatty acid oxidation disorders: Safety and efficacy in children and adults following 24weeks of treatment. Mol Genet Metab. 2017;120:370–7.
Gillingham MB, Heitner SB, Martin J, Rose S, Goldstein A, El-Gharbawy AH, et al. Triheptanoin versus trioctanoin for long-chain fatty acid oxidation disorders: a double blinded, randomized controlled trial. J Inherit Metab Dis. 2017;40:831–43.
Vockley J, Marsden D, McCracken E, DeWard S, Barone A, Hsu K, et al. Long-term major clinical outcomes in patients with long chain fatty acid oxidation disorders before and after transition to triheptanoin treatment--A retrospective chart review. Mol Genet Metab. 2015;116:53–60.
Tucci S, Floegel U, Beermann F, Behringer S, Spiekerkoetter U. Triheptanoin: long-term effects in the very long-chain acyl-CoA dehydrogenase-deficient mouse. J Lipid Res. 2017;58:196–207.
Djouadi F, Aubey F, Schlemmer D, Ruiter JP, Wanders RJ, Strauss AW, et al. Bezafibrate increases very-long-chain acyl-CoA dehydrogenase protein and mRNA expression in deficient fibroblasts and is a potential therapy for fatty acid oxidation disorders. Hum Mol Genet. 2005;14:2695–703.
Djouadi F, Bastin J. PPARs as therapeutic targets for correction of inborn mitochondrial fatty acid oxidation disorders. J Inherit Metab Dis. 2008;31:217–25.
Djouadi F, Habarou F, Le Bachelier C, Ferdinandusse S, Schlemmer D, Benoist JF, et al. Mitochondrial trifunctional protein deficiency in human cultured fibroblasts: effects of bezafibrate. J. Inherit. Metab. Dis. 2016;39:47–58.
Yamaguchi S, Li H, Purevsuren J, Yamada K, Furui M, Takahashi T, et al. Bezafibrate can be a new treatment option for mitochondrial fatty acid oxidation disorders: evaluation by in vitro probe acylcarnitine assay. Mol Genet Metab. 2012;107:87–91.
Li H, Fukuda S, Hasegawa Y, Kobayashi H, Purevsuren J, Mushimoto Y, et al. Effect of heat stress and bezafibrate on mitochondrial beta-oxidation: comparison between cultured cells from normal and mitochondrial fatty acid oxidation disorder children using in vitro probe acylcarnitine profiling assay. Brain Dev. 2010;32:362–70.
Bonnefont JP, Bastin J, Behin A, Djouadi F. Bezafibrate for an inborn mitochondrial beta-oxidation defect. N Engl J Med. 2009;360:838–40.
Bonnefont JP, Bastin J, Laforet P, Aubey F, Mogenet A, Romano S, et al. Long-term follow-up of bezafibrate treatment in patients with the myopathic form of carnitine palmitoyltransferase 2 deficiency. Clin Pharmacol Ther. 2010;88:101–8.
Yamada K, Shiraishi H, Oki E, Ishige M, Fukao T, Hamada Y, et al. Open-label clinical trial of bezafibrate treatment in patients with fatty acid oxidation disorders in Japan. Mol Genet Metab Rep. 2018;15:55–63.
Orngreen MC, Madsen KL, Preisler N, Andersen G, Vissing J, Laforet P. Bezafibrate in skeletal muscle fatty acid oxidation disorders: a randomized clinical trial. Neurology. 2014;82:607–13.
Bastin J, Bonnefont JP, Djouadi F, Bresson JL. Should the beneficial impact of bezafibrate on fatty acid oxidation disorders be questioned? J Inherit Metab Dis. 2015;38:371–2.
Orngreen MC, Vissing J, Laforet P. No effect of bezafibrate in patients with CPTII and VLCAD deficiencies. J Inherit Metab Dis. 2015;38:373–4.
Acknowledgements
We are grateful to Professors Seiji Yamaguchi and Seiji Fukuda, in the Department of Pediatrics of Shimane University, for their critical suggestions and comments on this article. This study was supported by JSPS KAKENHI (grant number 16K21179).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Yamada, K., Taketani, T. Management and diagnosis of mitochondrial fatty acid oxidation disorders: focus on very-long-chain acyl-CoA dehydrogenase deficiency. J Hum Genet 64, 73–85 (2019). https://doi.org/10.1038/s10038-018-0527-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s10038-018-0527-7
This article is cited by
-
ACADL-YAP axis activity in non-small cell lung cancer carcinogenicity
Cancer Cell International (2024)
-
Didymin protects pancreatic beta cells by enhancing mitochondrial function in high-fat diet-induced impaired glucose tolerance
Diabetology & Metabolic Syndrome (2024)
-
Structural basis for defective membrane targeting of mutant enzyme in human VLCAD deficiency
Nature Communications (2022)
-
Evidence that Oxidative Disbalance and Mitochondrial Dysfunction are Involved in the Pathophysiology of Fatty Acid Oxidation Disorders
Cellular and Molecular Neurobiology (2022)
-
Visceral obesity and insulin resistance associate with CD36 deletion in lymphatic endothelial cells
Nature Communications (2021)
