
Abstract
We present a case of a newborn female with multiple anomalies demonstrating that the causes of imprinting disorders rely not only on the parent-of-origin of the chromosomal aberrations, but also the scope of genes contained in the imprinted region. The newborn female presented with prenatal polyhydramnios, neonatal respiratory distress, distal contractures, abdominal hernia, bell-shaped thorax, and abnormal ribs. The neonate required mechanical ventilation due to apnea, underwent surgery for laryngomalacia, and showed development delay by age 11 months. Chromosomal microarray analysis identified a single copy number loss in chromosome region 14q32.2q32.31, containing genes that are differentially expressed based on parent-of-origin. Microarray analysis also confirmed the identical deletion in the patient’s mother, who was reported to be normal. Additional molecular analyses determined the exact size and breakpoints of the deletion as well as methylation states in both the patient and her mother. The maternally transmitted deletion was responsible for Kagami–Ogata syndrome in the patient.
Similar content being viewed by others


Introduction
Genomic imprinting is an epigenetic marking phenomenon that allows gene expression predominantly from a single parental allele [1,2,3]. Imprinted genes, located on a specific region of a chromosome, form a cluster with differentially methylated regions (DMRs). DMRs are methylated in only one of two alleles and regulate expression of imprinted genes in a parent-of-origin-dependent manner [2, 3]. The parent-of-origin-dependent gene expression in imprinted regions has been explained in association with various regulatory mechanisms [3, 4]. A frequently observed phenomenon is a gene expression inhibitory mechanism between paternally and maternally expressed genes [5]. It was recently reported that the human genome harbors more than 70 imprinting-associated DMRs [6].
The human 14q32 locus is a genomic imprinting region and consists of maternally expressed non-protein coding genes and paternally expressed protein-coding genes [7]. The maternally expressed genes (MEGs) include long noncoding RNAs (MEG3 and MEG8), RTL1 antisense (RTL1as), small nucleolar RNAs (snoRNAs), and a large group of microRNAs (miRNAs). The paternally expressed genes include DLK1 and RTL1.
There are two DMRs in the 14q32 imprinted region. The intergenic DMR (IG-DMR) is located between DLK1 and MEG3, and the MEG3-DMR is in the promoter region of MEG3 [7]. Maternally inherited DMRs are in an unmethylated state, whereas paternally inherited DMRs are in a methylated state. IG-DMR maintains the parent-of-origin-dependent methylation patterns in the body and placenta, while MEG3-DMR does so only in the body [8]. IG-DMR regulates the gene expression mainly in the placenta. Unmethylated MEG3-DMR regulates expression of MEGs in the body, and its methylation state is under the control of the IG-DMR. The methylation control between DMRs seems to be unidirectional, as the methylation state of IG-DMR is independent of that of MEG3-DMR [8].
Kagami–Ogata syndrome (KOS14, OMIM #608149) is caused by paternal uniparental disomy of chromosome 14 (UPD14), maternal deletion of the 14q32.3 imprinted region, or hypermethylation of IG-DMR and/or MEG3-DMR [7, 9]. In contrast, maternal UPD14, paternal deletion of the 14q32.2 imprinted region, or hypomethylation of DMRs cause Temple syndrome (TS14, OMIM #616222) [1, 10]. More than 50% of KOS14 and TS14 cases are caused by UPD14. Maternal deletions causing KOS14 and paternal deletions causing TS14 account for approximately 20 and 10% of each disorder, respectively [2]. Clinical findings of KOS14 include a bell-shaped thorax, coat-hanger ribs, abdominal and thoracic wall defects, growth retardation, and polyhydramnios [2, 7]. TS14 is characterized by prenatal and postnatal growth retardation, hypotonia, feeding difficulties in infancy, truncal obesity, scoliosis, and precocious puberty [1, 2].
Here, we report a female infant with KOS14 resulting from a maternally inherited 14q32.2q32.31 deletion and provide a clinical description of the prenatal and postnatal presentation. The exact size and location of this deletion are unique among the relatively small number of previously described KOS14-causing deletions and our detailed description of its molecular and clinical consequences add to a growing body of knowledge regarding this rare but possibly under-diagnosed condition.
Materials and methods
Chromosome microarray assay
Genomic DNA was extracted from peripheral whole blood using the EZ1 DNA Blood 350 µl Kit (Qiagen, Valencia, CA) with the EZ1 Advanced XL instrument (Qiagen, Valencia, CA) following the manufacturer’s instructions. Extracted DNA was quantified using the NanoDrop 1000 spectrophotometer (Thermo Fisher Scientific, Wilmington, DE). CMA was performed using the Affymetrix CytoScan® HD arrays following the laboratory procedures. Copy number variations (CNVs) were determined using the Chromosome Analysis Suite (ChAS) software (Affymetrix, Santa Clara, CA) with threshold of 50 probes for gain and of 25 probes for loss.
Breakpoint determination
PCR products were generated from genomic DNA samples of the patient and her mother using a Phusion high-fidelity DNA polymerase (New England BioLabs, Ipswich, MA) with UPD14-S1F (5′-ATGGTTTGTGATTTCATGGGTCTTG-3′) and UPD14-S1R (5′- TGGACTCAACATGTGTAGAAATGGA-3′) primers. DNA sequencing reactions were performed using the BigDye Terminator v3.1 Kit (Life Technologies Corporation, Grand Island, NY) with nested primers UPD14-S2F (5′-GAGACAGAAGTGGGAAGGGC-3′) and UDP14-S2R (5′-TGATTTGTTTATGTTATGACGCATGGA-3′). DNA sequences were obtained using the 3500 Genetic Analyzer (Applied Biosystems, Carlsbad, CA).
Methylation analysis
A combined bisulfite restriction analysis was performed following published procedures [8]. Briefly, genomic DNA was treated with bisulfite using EpiMark Bisulfite Conversion Kit (New England BioLabs, Ipswich, MA). PCR was performed using AmpliTaq Gold (Thermo Fisher Scientific, Foster City, CA) with published CG4, CG6, or CG7 primers [8]. PCR products were digested with Bst U I (for CG4 and CG7) or Taq I (for CG6) (New England BioLabs, Ipswich, MA) for 2 h and then separated on 2% E-Gel® EX agarose gels (Invitrogen by Thermo Fisher Scientific, Carlsbad, CA) with a 50-bp size ladder (New England BioLabs, Ipswich, MA). Control samples were normal diploid clinical samples, as determined in our laboratory by CMA analysis.
Deletion coordinates in KOS14 patients
Genome coordinates of genes in the chromosome 14 imprinted region and deletion positions of reported KOS14 patients are based on the hg19 assembly. Coordinates based on other assemblies were converted to hg19 using the Lift Genome Annotations tool (https://genome.ucsc.edu/cgi-bin/hgLiftOver).
Results
Clinical history
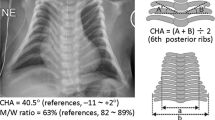
The maternal history was significant for gestational diabetes and pregnancy-induced hypertension. Prenatal ultrasounds indicated possible tracheoesophageal (TE) fistula due to polyhydramnios (AFI ~50) and small stomach bubble. Mild left ventriculomegaly and macroglossia were also reported. Induction of labor was initiated at 37 1/7 weeks gestation due to pregnancy-induced hypertension. After failure to progress, C-section delivery was completed. The gross pathological description of the 470 gram placenta was unremarkable. Neonatal chest radiograph showed small stomach bubble and abnormal appearance to the ribs (Fig. 1f). Given concern for TE fistula, patient was transferred to Intensive Care Nursery (ICN) for evaluation and management.

Clinical appearances (a–e) and chest radiograph (f) of the patient. Genetics physical exam noticed dysmorphic features including: (a) broad forehead, open mouth, and cupid’s bow to upper lip; (b) short upturned nose; (c) overlapping fingers; (d) transverse palmar crease on left hand; and e long toes. Patient’s chest radiography performed at one-month-old demonstrated bell-shaped thorax and coat-hanger ribs (f)
ICN admission examination noted silent cry, hypotonia, macroglossia, overlapping fingers and flexed wrists. Genetics consultation was requested due to fetal anomalies and, in addition to the above features, examination noted mild retrognathia, redundant anterior neck skin, camptodactyly, single transverse palmar crease on left, and long toes (especially 5th toes). Echocardiogram and head ultrasound were unremarkable. Brain MRI was essentially normal with findings consistent with prematurity or immature brain development. Physical and occupational therapies were initiated in the ICN and finger splints and stretching were initiated at about 1 week of age.
During her extended newborn admission, the patient experienced respiratory symptoms including central and obstructive apnea requiring occasional continuous positive airway pressure (CPAP). Laryngomalacia was noted by pediatric otolaryngology and endoscopic bilateral laser supraglottoplasty was performed at 1 month of age. The patient’s chest radiograph showed bell-shaped thorax and coat-hanger ribs (Fig. 1f). Re-evaluation by Genetics at 1 month of age identified resolution of the redundant neck skin. In addition to her other previously noted features, bitemporal narrowing, widened inner canthal distance, depressed nasal bridge, and retrognathia with mild macroglossia were reported. The patient was discharged at 5 weeks of age.
At 6 months of age, the patient was seen for outpatient Genetics re-evaluation. Developmentally, she was rolling, almost sitting, and beginning to crawl. The patient was normocephalic with length and weight at the 2-4th percentile. Parents reported feeding difficulties and esotropia of her right eye. Apneic episodes were decreasing, though desaturations were still regularly noted by overnight pulse oximetry but no interventions were required. Physical exam noted broad forehead, small palpebral fissures, widened inner canthal distance, short upturned nose, open mouth, cupid’s bow upper lip, and retrognathia (Fig. 1a, b). Her palmar creases were bridged and toes were overlapping (Figs. 1c–e). Finger contractures had resolved (Fig. 1d). The patient had narrow-appearing thorax and indented sternal notch. She had hypotonia, doughy muscles, as well as umbilical, abdominal, and inguinal hernias.
Physical examination at 1 year of age was largely unchanged. Feeding difficulties persisted but growth parameters were within normal limits. Thin upper lip, small palpebral fissures still with widened inner canthal distance, and soft skin were noted.
Developmental assessments were performed at 11 and approximately 18 months of age through Child Development Program. Assessments were completed using Bayley Scales of Infant and Child Development-III. At 11 months of age, cognitive and language scores (receptive and expressive) were equivalent to those of a 6–7 month old. Fine and gross motor skills corresponded to those of a 7–8 month old. At 17 months 25 days of age, her cognitive level was at an 11 month level. Receptive and expressive language skills were at 9 and 12 month levels, respectively. Gross and fine motor skills were at a 10 month level. The patient is receiving early intervention services.
Chromosome microarray analysis
To understand underlying genetic defects in the patient, we performed CMA as a constitutional assay. After conducting thorough analysis for pathogenic chromosome CNVs, a copy number loss in the long arm of chromosome 14 (14q32.2q32.31) was detected (Fig. 2a). It was a single copy deletion and the size of the deletion was approximately 133 kb. Genes contained within the deletion region are MIR2392, MEG3, MIR770, MIR493, MIR337, MIR665, RTL1, RTL1as, MIR431, MIR433, MIR127, MIR432, MIR136, MEG8, SNORD112, MIR370, SNORD113-1, SNORD113-2, SNORD113-3, SNORD113-4, SNORD113-5, SNORD113-6, SNORD113-7, and SNORD113-8. These genes are differentially expressed based on the parent-of-origin. No other pathogenic CNVs or long contiguous stretches of homozygosity were detected by CMA.

Molecular analysis of a maternally inherited deletion in 14q32 in the patient. (a) CMA with a CytoScan HD array determined copy number loss in the patient (presented in blue color) and the exact same deletion in the patient’s mother (in pink color). The top panel presents the segmented copy number state, the middle panel displays the weighted log2 ratio, and the bottom panel represents the copy number state. (b) Detailed analysis of the loss region found a gap between probes defining the proximal end of the loss region and probes calling a normal copy number of two. The gap is marked with dotted lines and the size is approximately 6 kb. Meanwhile, the distal end of the deletion region was well defined. (c) Exact breakpoints were determined by long and accurate PCR followed by the Sanger sequencing. Paternally inherited chromosome and expressed genes are displayed with blue color. Maternal counterparts are displayed with red color. Both DMRs are presented using gray bars. (d–f). The combined bisulfite restriction analysis was performed to determine methylation states of the CG4 (d) and CG6 (e) regions for IG-DMR, and of the CG7 (f) region for MEG3-DMR [8]. Restriction digested products were from methylated alleles in the CG4 (d) and CG7 (f) regions, and from unmethylated alleles in the CG6 (e) region. Lanes 1 and 7: the patient; lanes 2 and 8: the patient’s mother; lanes 3 and 9: normal control A; lanes 4 and 10: normal control B; lane 5: no template control. Lane 1 through 5 are PCR products, lane 7 through 10 are products of restriction digestion, and lanes M and 6 are a 50-bp size ladder
To understand the origin of the deletion, CMA testing, following the same protocol, was completed in the proband’s mother. The patient’s mother has an identical deletion in the chromosome 14 region (Fig. 2a), establishing maternal inheritance with the proband’s CMA result defined as arr[hg19] 14q32.2q32.31(101,278,207-101,410,792)x1 mat. The patient’s phenotype and CMA results are consistent with KOS14 (Table 1).
Breakpoint of the deleted region
The region of loss was declared by the ChAS software based on probe signal intensities compared to those of a reference group. Probe coverage appeared to be sufficient to call the deletion; however, the coverage was sparse in the proximal region of the deletion, while the coverage was dense in the distal end of the deletion (Fig. 2b). There was an approximately 6 kb gap between the software-declared loss region and the next available probe position. Detailed sequences analysis found that IG-DMR locates within the gap so that we were unable to discern whether the patient’s loss region harbors these two DMRs or only MEG3-DMR.
We determined the exact breakpoints in the deletion using long and accurate PCR followed by Sanger sequencing of PCR products. The proximal breakpoint of the deletion was between IG-DMR and MEG3-DMR, and the distal breakpoint was between SNORD113-8 and SNORD113-9 (Fig. 2c). Genomic coordinates of the breakpoints were chr14:101277971-101410566 (hg19), and the exact size of the deletion was 132596 bp, almost identical to the size provided by the analysis software. These results indicate that the patient still harbored IG-DMRs inherited from both parents and that she had only a paternally inherited MEG3-DMR due to the maternal deletion.
Methylation states of IG-DMR and MEG3-DMR
We speculated that the patient may have a methylated and an unmethylated IG-DMR, on her paternal and maternal copies of chromosome 14, respectively, and that the patient may harbor only methylated MEG3-DMR because of the loss of maternally inherited unmethylated MEG3-DMR. In contrast, the proband’s mother may have only unmethylated (maternal) MEG3-DMR that might cause TS14. To prove our hypothesis, we performed the combined bisulfite restriction analysis, which determined methylation states of the CG4 and CG6 regions for IG-DMR and of the CG7 region for MEG3-DMR (Fig. 2d–f). The patient and her mother generated both methylated and unmethylated IG-DMRs in approximately equimolar amounts, as anticipated (Fig. 2d, e). Meanwhile, the patient produced only methylated MEG3-DMR determined by the methylation state in CG7, while the patient’s mother produced only unmethylated MEG3-DMR (Fig. 2f), indicating loss of the paternal MEG3-DMR. These results were concordant with our speculation based on results of the breakpoint assay.
Discussion
This child has been diagnosed as having KOS14 due to a maternally inherited single copy deletion on 14q32.2. The patient was noted to have multiple prenatal and postnatal abnormalities, and CMA determined the cause and origin of the patient’s KOS14 phenotype. Molecular biological assays detected affected imprinted genes and regions, and provided confirmatory evidence for KOS14. The assays also determined that the single copy deletion on the paternal chromosome was insufficient to cause an obvious TS14 phenotype in the patient’s mother.
According to our survey of the current medical literature, this patient is only the seventh reported case of KOS14 with deletion only in MEG3-DMR with intact IG-DMR (Fig. 3) [8, 11, 12]. In spite of a deletion in MEG3-DMR, the methylation states in IG-DMR were indistinguishable from those of wild-type controls (Fig. 2d–f). The presence of two distinct methylation states of IG-DMR even in the absence of MEG3-DMR supported the unidirectional hierarchical relationship between these two DMRs, in which IG-DMR governs the methylation states of MEG3-DMR, but not the other way around [8, 11].

Genomic locations of deletions that have caused Kagami–Ogata syndrome (KOS14). Paternally inherited chromosome and expressed genes are displayed with blue line and blue-filled boxes, respectively. Maternally inherited chromosome and expressed genes are presented with red color. Gray bars represent positions of IG-DMR and MEG3-DMR. Black bars localize loss regions in KOS14 patients. The scale bar corresponds to 50 kb. Genomic coordinates of these genes and loss regions are of the GRCh37 (hg19) assembly (Supplemental Information Table 1). Deletion 1: Kagami 2008 [9] (patients 1 and 2); Deletion 2: Kagami 2008 [9] (patient 3); Deletion 3: Kagami 2008 [9] (patient 4); Deletion 4: Kagami 2008 [9] (patient 5); Deletion 5: Kagami 2010 [8] (patient 1); Deletion 6: Kagami 2010 [8] (patient 2); Deletion 7: Beygo 2015 [11] (patient 1); Deletion 8: Beygo 2015 [11] (patients 2 and 3); Deletion 9: Corsello 2015 [15]; Deletion 10: Rosenfeld 2015 [14] (patient 4); Deletion 11: Rosenfeld 2015 [14] (patient 5); Deletion 12: van der Wef 2016 [12] (patients AII.1 and AII.2); Deletion 13: van der Werf 2016 [12] (patients BII.1 and BII.3)
So far there have been no reported cases of KOS14 or TS14 caused by mutation of a single imprinted gene [1]. Studies of patients with rare paternal deletions in the region have determined that DLK1 and MEG3, including IG-DMR and MEG3-DMR, are the responsible regions for TS14 [9]. Our study demonstrated that the patient’s mother harbors intact DLK1, and normal methylation patterns in IG-DMR (Fig. 2d, e); however, her MEG3-DMR is in an unmethylated state (Fig. 2f). The patient’s mother appears to be clinically normal, with no features to suggest TS14 [10]. These results indicate that the MEG3-DMR hypomethylation state may be unrelated to TS14, and that responsible regions for the syndrome could be further defined to DLK1 and IG-DMR. Our results are consistent with recently performed genotype-phenotype studies of TS14 [8, 13].
Two plausible mechanisms of KOS14 have been proposed. RTL1 overexpression by the loss of RTL1as (with or without loss of other MEGs) has been proposed for KOS14 phenotypes [7, 9]. This idea was supported by KOS14 patients with 14q32.2 deletions affecting RTL1as but not any of the DMRs [14, 15]. Secondly, after failing to find a commonly deleted region among newly reported deletion cases (Fig. 3), another group suggests that multiple redundant elements in the region may be involved in KOS14 phenotypes; therefore, KOS14 may be caused by abnormal expression of any combination of MEGs either by disruption of DMR regulatory function, or by physical deletion in some of these genes [12]. Additional case studies and investigation are required to establish a reliable genotype-phenotype relationship for KOS14 prior to determining responsible element(s) and regulation mechanisms. The diagnosis of KOS14 in our patient due to the deletion on 14q32.2 will contribute to this endeavor.
More attention must be paid to miRNAs and snoRNAs present in the region because of their critical biological functions [5, 16,17,18]. One of the largest human miRNA gene clusters locates in this imprinted region [17]. There are also two groups of snoRNAs in the region. Their roles in the KOS14 phenotypes, however, have barely been investigated [8]. The SNORD116, another group of snoRNAs on chromosome 15, for instance, emerged as a responsible element for Prader-Willi syndrome, another imprinting disorder [19]. Investigating expression levels of miRNAs and snoRNAs in different KOS14 deletion patients would provide initiatives for determining their roles in KOS14 phenotypes. It would also be critical for monitoring of other diseases as well because aberration of these RNA molecules seems to be pathogenic [17].
In our patient, the absence of maternal gene expression and/or enhanced paternal gene expression may be responsible for the development of the patient’s abnormalities. Candidate genes are maternally expressed MEG3, RTL1as, MEG8, and some of the snoRNAs, which locate within the deleted region (Fig. 3). The rest of the snoRNAs and miRNAs could also be causative genes (Fig. 3) because their expression is under the control of hypomethylated MEG3-DMR, which is absent in the patient (Fig. 2f) [20, 21]. Among the paternal genes, RTL1 is likely overexpressed in the patient, as observed in patients with the RTL1as deletion, and the RTL1 overexpression might cause the patient’s abnormalities [8, 9, 22]. Expression of maternal DLK1 was observed in a patient with an MEG3-DMR deletion [8], so increased expression of DLK1 could also be a candidate causative gene, though its contribution seems to be minimal [7]. It is possible that a combination of the absence of maternal gene expression and enhanced paternal gene expression causes the patient’s clinical features. The identity of the causative gene(s), however, remains to be determined.
IG-DMR and MEG3-DMR may play distinct roles in the maternal gene expression: IG-DMR may affect expression of genes in a wide chromosomal region, whereas MEG3-DMR may control expression of a specific group of genes within the region. The hierarchical relationship between IG-DMR and MEG3-DMR in the body [8, 11] supports this idea. In mouse embryonic stem cells, it was demonstrated that ZFP57, a zinc finger transcription factor, recognizes a methylated motif and recruits histone modification enzymes to generate the chromatin region in a repressive state [23]. It is plausible that human IG-DMR has a similar functional motif for parent-of-origin-dependent methylation patterns in the region. When the chromatin region is in an active chromatin state, it might play the role of an enhancer for MEG3 [20]. The activated enhancer, however, might require additional factor(s). In mouse embryonic stem cells, AFF3, a transcription component, was recruited to the enhancer to induce MEG3 expression [24]. In humans, the transcriptional regulator CCCTC-binding factor (CTCF) may be involved in maternal gene expression because CTCF binding sites were found around MEG3-DMR [8, 25] and because MEG3 was downregulated in CTCF knockdown mice [26]. Recently, however, RNA polymerase II, but not CTCF, was shown to bind to the intergenic/intronic hypomethylated regions (iHMR) located upstream of MEG3-DMR in mouse embryonic fibroblast cells [27]. Isolation of the chromatin regulatory motif in IG-DMR and iHMR in the upstream region of MEG3-DMR needs to be investigated to understand the detailed parent-of-origin-dependent gene expression mechanisms in humans.
At the time this manuscript is being written the patient is a healthy two year old. In spite of her global developmental delay, the patient’s progress is quite impressive. Although KOS14 phenotypes are quite variable [14], survival rates of KOS14 caused by microdeletion were estimated to be 50% after the age of five [28]. This patient could be another example of a long-surviving case of KOS14 caused by deletion [12]. Despite possible ascertainment bias in the current reported literature, management of her other symptoms will be critical as two of three KOS14 microdeletion patients died of a respiratory infection [28] and our patient still requires occasional mechanical ventilation (Table 1).
Molecular diagnostics was critical to determine the genetic defect and diagnosis in this patient. The chest radiography obviously demonstrated typical KOS14 findings such as the bell-shaped thorax and coat-hanger ribs in the patient (Fig. 1f and Table 1); however, it was not until the CMA results were available that the patient’s constellation of findings were noted to be consistent with KOS14. A diagnostic flow chart [7] suggests that if the chest findings were more carefully examined, a methylation analysis would be the first test followed by parent-of-origin and deletion analysis. Given the low incidence of KOS14, this diagnosis may not be immediately recognized and may only be suspected following CMA analysis. In cases where KOS14 is clinically suspected, simultaneous CMA and methylation analysis would be an ideal strategy to achieve the shortest time to diagnosis. The molecular analysis findings would also be essential for genetic counseling. In this case, genetic counseling and prenatal genetic diagnosis for future pregnancies is warranted due to the 50% recurrence risk for the proband’s mother to transmit the deletion to future children.
References
Eggermann T, Perez de Nanclares G, Maher ER, Temple IK, Tümer Z, Monk D, et al. Imprinting disorders: a group of congenital disorders with overlapping patterns of molecular changes affecting imprinted loci. Clin Epigenetics. 2015;7:123.
Soellner L, Begemann M, Mackay DJ, Grønskov K, Tümer Z, Maher ER, et al. Recent Advances in Imprinting Disorders. Clin Genet. 2017;91:3–13.
Reik W, Walter J. Genomic imprinting: parental influence on the genome. Nat Rev Genet. 2001;2:21–32.
Hnisz D, Day DS, Young RA. Insulated neighborhoods: structural and functional units of mammalian gene control. Cell. 2016;167:1188–1200.
Seitz H, Youngson N, Lin SP, Dalbert S, Paulsen M, Bachellerie JP, et al. Imprinted microRNA genes transcribed antisense to a reciprocally imprinted retrotransposon-like gene. Nat Genet. 2003;34:261–2.
Joshi RS, Garg P, Zaitlen N, Lappalainen T, Watson CT, Azam N, et al. DNA methylation profiling of uniparental disomy subjects provides a map of parental epigenetic bias in the human genome. Am J Hum Genet. 2016;99:555–66.
Ogata T, Kagami M. Kagami–Ogata syndrome: a clinically recognizable upd(14)pat and related disorder affecting the chromosome 14q32.2 imprinted region. J Hum Genet. 2016;61:87–94.
Kagami M, O’Sullivan MJ, Green AJ, Watabe Y, Arisaka O, Masawa N, et al. The IG-DMR and the MEG3-DMR at human chromosome 14q32.2: hierarchical interaction and distinct functional properties as imprinting control centers. PLoS Genet. 2010;6:e1000992.
Kagami M, Sekita Y, Nishimura G, Irie M, Kato F, Okada M, et al. Deletions and epimutations affecting the human 14q32.2 imprinted region in individuals with paternal and maternal upd(14)-like phenotypes. Nat Genet. 2008;40:237–42.
Ioannides Y, Lokulo-Sodipe K, Mackay DJ, Davies JH, Temple IK. Temple syndrome: improving the recognition of an underdiagnosed chromosome 14 imprinting disorder: an analysis of 51 published cases. J Med Genet. 2014;51:495–501.
Beygo J, Elbracht M, de Groot K, Begemann M, Kanber D, Platzer K, et al. Novel deletions affecting the MEG3-DMR provide further evidence for a hierarchical regulation of imprinting in 14q32. Eur J Hum Genet. 2015;23:180–8.
van der Werf IM, Buiting K, Czeschik C, Reyniers E, Vandeweyer G, Vanhaesebrouck P, et al. Novel microdeletions on chromosome 14q32.2 suggest a potential role for non-coding RNAs in Kagami–Ogata syndrome. Eur J Hum Genet. 2016;24:1724–9.
Severi G, Bernardini L, Briuglia S, Bigoni S, Buldrini B, Magini P, et al. New patients with Temple syndrome caused by 14q32 deletion: genotype-phenotype correlations and risk of thyroid cancer. Am J Med Genet A. 2016;170A:162–9.
Rosenfeld JA, Fox JE, Descartes M, Brewer F, Stroud T, Gorski J,L, et al. Clinical features associated with copy number variations of the 14q32 imprinted gene cluster. Am J Med Genet A. 2015;167A:345–53.
Corsello G, Salzano E, Vecchio D, Antona V, Grasso M, Malacarne M, et al. Paternal uniparental disomy chromosome 14-like syndrome due a maternal de novo 160 kb deletion at the 14q32.2 region not encompassing the IG- and the MEG3-DMRs: Patient report and genotype-phenotype correlation. Am J Med Genet A. 2015;167A:3130–8.
Kanduri C. Long noncoding RNAs: lessons from genomic imprinting. Biochim Biophys Acta. 2016;1859:102–11.
Benetatos L, Hatzimichael E, Londin E, Vartholomatos G, Loher P, Rigoutsos I, et al. The microRNAs within the DLK1-DIO3 genomic region: involvement in disease pathogenesis. Cell Mol Life Sci. 2013;70:795–814.
de Almeida RA, Fraczek MG, Parker S, Delneri D, O’Keefe RT. Non-coding RNAs and disease: the classical ncRNAs make a comeback. Biochem Soc Trans. 2016;44:1073–8.
Sahoo T, del Gaudio D, German JR, Shinawi M, Peters SU, Person RE, et al. Prader-Willi phenotype caused by paternal deficiency for the HBII-85 C/D box small nucleolar RNA cluster. Nat Genet. 2008;40:719–21.
Enterina JR, Enfield KSS, Anderson C, Marshall EA, Ng KW, Lam WL. DLK1-DIO3 imprinted locus deregulation in development, respiratory disease, and cancer. Expert Rev Respir Med. 2017;11:749–61.
da Rocha ST, Edwards CA, Ito M, Ogata T, Ferguson-Smith AC. Genomic imprinting at the mammalian Dlk1-Dio3 domain. Trends Genet. 2008;24:306–16.
Kagami M, Matsuoka K, Nagai T, Yamanaka M, Kurosawa K, Suzumori N, et al. Paternal uniparental disomy 14 and related disorders: placental gene expression analyses and histological examinations. Epigenetics. 2012;7:1142–50.
Quenneville S, Verde G, Corsinotti A, Kapopoulou A, Jakobsson J, Offner S, et al. In embryonic stem cells, ZFP57/KAP1 recognize a methylated hexanucleotide to affect chromatin and DNA methylation of imprinting control regions. Mol Cell. 2011;44:361–72.
Wang Y, Shen Y, Dai Q, Yang Q, Zhang Y, Wang X, et al. A permissive chromatin state regulated by ZFP281-AFF3 in controlling the imprinted Meg3 polycistron. Nucleic Acids Res. 2017;45:1177–85.
Wylie AA, Murphy SK, Orton TC, Jirtle RL. Novel imprinted DLK1/GTL2 domain on human chromosome 14 contains motifs that mimic those implicated in IGF2/H19 regulation. Genome Res. 2000;10:1711–8.
Wan LB, Pan H, Hannenhalli S, Cheng Y, Ma J, Fedoriw A, et al. Maternal depletion of CTCF reveals multiple functions during oocyte and preimplantation embryo development. Development. 2008;135:2729–38.
Bakshi A, Bretz CL, Cain TL, Kim J. Intergenic and intronic DNA hypomethylated regions as putative regulators of imprinted domains. Epigenomics. 2018;10:445–61.
Kagami M, Kurosawa K, Miyazaki O, Ishino F, Matsuoka K, Ogata T. Comprehensive clinical studies in 34 patients with molecularly defined UPD(14)pat and related conditions (Kagami–Ogata syndrome). Eur J Hum Genet. 2015;23:1488–98.
Acknowledgements
The authors wish to thank the staff of the Laboratory for Clinical Genomics and Advanced Technology (CGAT). The data presented in this manuscript was generated through CGAT in the Department of Pathology and Laboratory Medicine of the Geisel School of Medicine at Dartmouth, the Dartmouth Hitchcock Medical Center and the Norris Cotton Cancer Center. We would also like to thank Dr. Masayo Kagami for providing methylated and unmethylated genomic DNA sequence information of the CG4, CG6, and CG7 regions on 14q32.2 and coordinates of deletion cases.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Jung, HS., Vallee, S.E., Dinulos, M.B. et al. Maternally inherited 133kb deletion of 14q32 causing Kagami–Ogata syndrome. J Hum Genet 63, 1231–1239 (2018). https://doi.org/10.1038/s10038-018-0506-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s10038-018-0506-z
