
Abstract
Many features are shared between esophageal cancer (EC) and gastric cancer (GC). This study aimed to explore whether known EC susceptibility loci are also important in the development of GC. A total of 21 genetic variants associated with EC in genome-wide association studies were evaluated with association of GC risk in 2631 cases and 4373 controls of Chinese ancestry. Single variant and weighted genetic scores (WGS) for esophageal squamous cell carcinoma (ESCC), esophageal adenocarcinoma (EAC), and overall EC were analyzed with GC risk, respectively. Genetic variants of rs2274223 in PLCE1 at 10q23.33 (per G allele: odds ratio (OR) = 1.26, 95% confidence interval (CI): 1.16–1.38, P = 6.51 × 10−8), rs10052657 in PDE4D at 5q11.2 (per C allele: OR = 1.12, 95% CI: 1.01–1.25, P = 3.28 × 10−2) and rs671 in ALDH2 at 12q24.12 (per A-allele: OR = 0.83, 95% CI: 0.75–0.91, P = 1.14 × 10−4) were significantly associated with GC risk. The combined effect of those three variants had stronger influence on GC risk (OR = 1.31, 95% CI: 1.19–1.44, P = 2.34 × 10−8). High WGS of ESCC was also associated with increased risk of GC (P = 5.52 × 10−4 as a continuous variable) (trend test P = 2.71 × 10−4), whereas no statistically significant associations were observed between the WGS of EAC and GC risk (P = 0.66 as a continuous variable) (trend test P = 0.70). ESCC rather than EAC may share genetic susceptibility with GC. Genetic variants at 10q23.33, 5q11.2, and 12q24.12 may be useful as biomarkers to identify individuals with high risk for both ESCC and GC.
Similar content being viewed by others

Introduction
Esophageal cancer (EC) and gastric cancer (GC) are two common gastrointestinal cancers worldwide, with 456,000 new cases and 400,000 estimated deaths per year for EC, and 951,000 new cases and 723,000 estimated deaths per year for GC, respectively [1]. The stomach is connected to the esophagus through gastroesophageal junction, which is also known as cardia. Based on the anatomical location, GC can be classified into two types: true gastric (non-cardia) and gastroesophageal junction cancers (cardia) [2]. The majority of GC is gastric adenocarcinoma, whereas EC consists of two histopathological types, esophageal squamous cell carcinoma (ESCC) and esophageal adenocarcinoma (EAC) [3]. ESCC is predominant around the world especially in China, whereas EAC subtype is a major type in the United States, Australia, the United Kingdom, and other European countries [4].
Both environmental and genetic factors contribute to the development of EC and GC. There are similarities and differences in the risk factors for ESCC and EAC. Tobacco use was associated with increased risk of both ESCC and EAC [5,6,7]. Alcohol consumption is a specific risk factor for ESCC [8, 9], whereas gastroesophageal reflux disease, obesity, and Barrett’s esophagus were associated with increased susceptibility of EAC [9]. The known environmental risk factors for GC are Helicobacter pylori infection, smoking, obesity, low intake of fresh fruits and vegetables, and high consumption of salted foods [2]. Genome-wide association studies (GWAS) have been conducted to explore genetic variants influencing the susceptibility of EC and GC over the past few decades [10,11,12]. A missense mutation located in PLCE1 named rs2274223 was found to be associated with risk of ESCC and gastric cardia cancer [10].
In consideration of the close anatomical location and similarities among risk factors between EC and GC, we hypothesized that the genetic basis of developing EC and GC might have something in common. To test whether single EC risk variant or cumulative genetic risk score computed using established EC risk loci were also associated with GC risk, we utilized risk loci reported in the published EC GWAS and tested whether they were associated with GC in our large case–control studies.
Materials and methods
Study populations
Participants of the current study were from three published GC GWAS. The Nanjing GWAS (565 cases and 1162 controls) and the Beijing GWAS (468 cases and 1123 controls) were based on two independent case–control studies, which were reported previously [12]. For the National Cancer Institute (NCI) GWAS (1625 cases and 2100 controls), subjects were recruited from Shanxi and Linxian [10]. All cases in the Nanjing and Beijing GWAS were diagnosed with non-cardia GC, whereas cases in the NCI GWAS contained both cardiac and non-cardia GC. In total, 2631 GC cases and 4373 controls were included in the current study. Basic demographic information of the participants was shown in Supplementary table 1.
Genetic variants selection
We searched the GWAS catalog (https://www.genome.gov/gwastudies/, last accessed on 25 July 2017) for genetic variants associated with EC risk. Besides, we searched PubMed database (https://www.ncbi.nlm.nih.gov/pubmed/) for recently published EC GWAS. The reported EC risk loci were filtered using the following criteria: (1) the reported significance level of the association reaching 5.00 × 10−8; (2) the minor allele frequency (MAF) of variants not < 1% in the Chinese population (1000 Genomes phase III); (3) for variants in linkage disequilibrium (LD) defined as r2 > 0.1, we selected the variant with the lowest P value. Finally, nine GWAS (eight studies for ESCC, one study for EAC) were included in our current study [10, 11, 13,14,15,16,17,18,19]. 42 single nucleotide polymorphisms (SNP) reached the predefined significance level of association, but four had the MAF of < 1% in the Chinese population. After excluding SNPs in LD, 21 SNPs (18 SNPs for ESCC, 3 SNPs for EAC) were included in the final statistical analysis. Detailed information about the eligible EC risk loci are shown in Supplementary table 2.
Imputation and quality control
After basic quality–control procedures performed in GWAS, we excluded SNPs with call rate < 95%, MAF < 0.01, and Hardy–Weinberg equilibrium P value < 1.00 × 10−6. Then, we performed imputation for the Nanjing (Affymetrix 6.0), the Beijing (Affymetrix 6.0) and the NCI (Illumina 660 W) GWAS separately using software SHAPEIT [20] and IMPUTE2 [21]. We used all populations from the 1000 Genomes Project Phase III as the reference set. After imputation, we further excluded SNPs with poor imputation quality (info score < 0.3) and repeated the quality control procedures for SNPs mentioned above. Among the 21 selected SNPs, we did not get genotype information of rs76014404 and rs8030672 from the Nanjing/Beijing or the NCI GWAS. Therefore, we used two SNPs (rs2143771 and rs116760846) in complete LD with these SNPs in the following analyses (Supplementary table 3).
Calculation of weighted genetic scores
Weighted genetic score (WGS) was calculated to evaluate the cumulative effect of esophagus cancer risk loci on GC risk. We calculated two independent WGS for ESCC and EAC, as there was high heterogeneity in the genetic background between these two subtypes. We also combined the two subtypes to measure the WGS for overall EC. For each individual, WGS was calculated by multiplying the number of risk alleles by the EC-associated beta (βj), which was derived from published studies. For rs2274223, which was reported in more than one study, we estimated its effect on EC based on meta-analysis. To calculate WGS for the ith subject, the following formula was used:
In this formula, xij is the number of risk alleles for the j-th variant in the i-th subject (xij=0, 1, or 2) and βj is the coefficient or weight for the j-th variant (calculated by ln-transformed of odds ratios (ORs) from published studies).
Differential expression analysis
Expression data (normalized expectation-maximization read counts) were downloaded from the Cancer Genome Atlas database, which consisted of 87 EAC tissues, 10 EAC paired normal tissues, 85 ESCC tissues, three ESCC-paired normal tissues, 413 GC tissues, and 32 GC paired normal tissues, respectively. Expression data were log2 transformed to correspond to normal distribution. Paired t test (10 EAC tissues vs. 10 EAC paired normal tissues, 3 ESCC tissues vs. 3 ESCC paired normal tissues, 32 GC tissues vs. 32 GC paired normal tissues) and two-sample t test (87 EAC tissues vs. 10 EAC paired normal tissues, 85 ESCC tissues vs. 3 ESCC paired normal tissues, 413 GC tissues vs. 32 GC paired normal tissues) were used to evaluate differential expression among tumor and normal tissues.
Statistical analysis
Genetic association analysis was conducted by using logistic regression models. When dealing with association between single locus and GC risk, we assumed an additive genetic model in logistic regression. For GC risk-associated variants, we estimated the cumulative effect based on the number of risk alleles. We included the WGS in the logistic regression model both as a continuous variable and categorical variable. For the Nanjing and the Beijing GWAS, we adjusted for age, sex, smoking, drinking status, and principal component analysis (PCA) for population stratification, and for the NCI GWAS, we adjusted for age, sex, and PCA in the regression models. Meta-analysis was used to combine results from the three GWAS, and Cochran’s Q was used for heterogeneity test. Fixed-effect model was applied to assume the combined effect, whereas random effect model was repeated if I2 (calculated by 100% × (Q–(n–1))/Q) was > 75%. Differential expression analysis was performed based on two-sample t test or paired t test. Analyses were performed with Stata version 11 or R version 3.2.1, unless otherwise noted.
Results
Association between single variant and GC risk
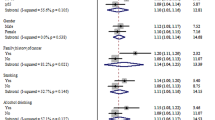
As shown in Table 1, among the 21 known genetic variants associated with EC risk, three were significantly associated with GC risk. Consistent with the previous report [10], the G allele of rs2274223 at 10q23.33 (reported gene: PLCE1) was associated with increased GC risk (OR = 1.26, 95% confidence interval (CI):1.16–1.38, P = 6.51 × 10−8). Considering that PLCE1 locus was identified as a common locus between ESCC and GC in the previous GWAS using NCI samples, we reanalyzed the association between rs2274223 and GC risk by excluding samples from NCI and found the direction of the association was consistent but not significant (per G allele: OR = 1.07, 95% CI: 0.94–1.23, P = 0.31). In addition, rs10052657 at 5q11.2 (reported gene: PDE4D) and rs671 at 12q24.12 (reported gene: ALDH2) were also associated with GC risk (OR = 1.12, 95% CI: 1.01–1.25, P = 3.28 × 10−2; OR = 0.83, 95% CI: 0.75–0.91, P = 1.14 × 10−4, respectively). Nevertheless, there were obvious heterogeneities among three studies for all the three discovered SNPs, and they were insignificant when random effect model was used in meta-analysis. We did not find significant associations with GC risk for the remaining 18 variants.
We further examined the cumulative effect of these three variants (rs2274223, rs10052657, and rs671) on GC risk (Table 2). We found a strong tendency of increased GC risk with greater numbers of risk alleles (OR = 1.31, 95% CI: 1.19–1.44, P = 2.34 × 10−8).
Association of EC WGS with GC risk
Because the observed effect of rs671 on ESCC and GC was in opposite directions, we derived the WGS based on the reported effect size of the 20 variants (excluding rs671) from the original EC study (Supplementary table 1) and evaluated the association of EC WGS and risk of GC (Table 3 and Table 4). We found that the EC WGS was significantly associated with increased risk of GC (OR = 1.15, 95% CI: 1.06–1.25, P = 1.20 × 10−3 for continuous WGS and OR = 1.08, 95% CI: 1.03–1.13, P = 9.11 × 10−4 for trend test for WGS categories).This association was mainly restricted to ESCC (OR = 1.16, 95% CI: 1.07–1.27, P = 5.52 × 10−4 for continuous WGS and OR = 1.09, 95% CI: 1.04–1.14, P = 2.71 × 10−4 for trend test for WGS categories) but not to EAC (OR = 1.02, 95% CI: 0.92–1.13, P = 0.66 for continuous WGS and OR = 0.99, 95% CI: 0.95–1.04, P = 0.70 for trend test for WGS categories).
Differential expression analysis of candidate genes
We further analyzed whether the expression levels of the genes associated with those three variants (rs2274223, rs10052657, and rs671) were altered in cancer tissues compared with normal tissues of esophagus and stomach (Supplementary figure 1). We found that PDE4D and ALDH2 were downregulated in both ESCC and GC tissues as compared with normal tissues. However, we did not observe differential expression for PLCE1 in either ESCC or GC tissues.
Discussion
In the current study, we investigated whether the known EC risk loci were associated with GC risk using 2631 GC cases and 4373 controls of Chinese ancestry. We found that the G allele of rs2274223, C allele of rs10052657, and G allele of rs671 were associated with increased risk of GC, and higher WGS of ESCC was associated with increased risk of GC.
PLCE1 is located on chromosome 10q23, which encodes a phospholipase enzyme that catalyzes the hydrolysis of phosphatidylinositol-4,5-bisphosphate to generate two second messengers: inositol 1,4,5-triphosphate and diacylglycerol [22]. In addition, it also interacts with small monomeric GTPases of the Ras and Rho families and heterotrimeric G proteins [23]. Thus, PLCE1 regulates various processes affecting cell growth, survival, differentiation, gene expression, and oncogenesis. Several studies found that the missense variation of rs2274223 in PLCE1 was significantly associated with ESCC [24] and gastric cardia cancer [25], but not EAC [26], which is consistent with our findings.
Rs10052657 was another locus significantly associated with GC. However, no additional study reported the association between rs10052657 and the risk of GC and EC. Rs10052657 is located in intron 5 of PDE4D, a gene that hydrolyzes the second messenger cAMP (cyclic adenosine monophosphate) and acts as a signal transduction molecule in multiple cell types. Previous studies have identified genetic variants in PDE4D were associated with risk of several cancers including breast cancer [27]. PDE4D was also found to be a diver gene participated in the development of cancer, and involved in cancer progression by accelerating proliferation [28,29,30]. PDE4D was overexpressed in prostate cancer [30, 31], whereas some PDE4D isoforms were downregulated [32, 33]. In our study, we observed down-regulation of PDE4D in both ESCC and GC tissues. These findings support the biological plausibility that genetic variants in PDE4D may confer altered risk to ESCC and GC, whereas the potential mechanism may involve different PDE4D isoforms as reported in studies of prostate cancer.
Unlike rs10052657, whose C allele increased risk of the above two cancers, A-allele of rs671 promoted ESCC but protected from GC. The SNP rs671 is located in the twelfth exon of ALDH2 at 12q24.12. ALDH2 belongs to the aldehyde dehydrogenase family and participates in pathway of alcohol metabolism. Several studies have shown that ALDH2 was associated with susceptibility to cancers including GC [34, 35], head and neck cancer [36], and colorectal cancer [37]. Rs671 has been reported to be associated with ESCC [38, 39], and overall EC [40], but the conclusions were inconsistent. Rs671 was also reported to influence GC risk, though their findings were opposite to ours [41, 42]. Alcohol consumption and rs671 were considered simultaneously when evaluating their associations with GC risk in those studies. Recently, one study reported that rs671 may not increase gastric cardia adenocarcinoma (GCA) susceptibility in Chinese Han populations, but the proportion of ALDH2 mutated allele carriers in GCA high-incidence areas was lower than that in low-incidence areas [43]. The above evidence suggested that the mutated allele may have a potential role in decreasing GC risk. In addition, Subjects with mutated ALDH2 exhibited a lower level of alcohol consumption than wild ALDH2 carriers [44]. Alcohol consumption is an established risk factor for cancers [45, 46] and healthy lifestyle like controlling alcohol intake is benefit for keeping cancers away. In conclusion, effect of rs671 on GC might depend on consumption of alcohol and A-allele may turn from a protective role to hazardous role in GC if subjects intake high level of alcohol.
From the analysis based on WGS, we observed a significant association between GC risk and WGS of ESCC rather than EAC. It suggested that ESCC and GC may share common genetic background. ESCC is the main histological type of EC in Asian countries, whereas the incidence of EAC now exceeds ESCC in European and American countries. There are great differences between the two histological types of EC in pathophysiology and pathogenesis [4]. However, most ESCC associated loci were discovered based on participants form Asian. In consideration of the prevalence of ESCC and GC in China, and the well-known shared genetic variant rs2274223 between the two cancers, there may be other genetic loci participate in occurrence of both cancers. Therefore, it is rational and credible to discover the shared genetic background between ESCC and GC. Although we did not find connections between EAC and GC, it may be explained by population heterogeneity as EAC associated loci were reported in European ancestry. Moreover, only a few EAC risk loci were reported and used in the current study, which might be less representative for genetic risk of EAC based on WGS of EAC.
There are some limitations in our study. First, although the association between ESCC WGS and GC risk was significant, there were obvious heterogeneities among three studies for three SNPs. The associations became weak as considering the heterogeneity and should be treated in caution. Second, we did not conduct subgroup analysis based on cardiac and non-cardiac GC, which limited our analysis on the different impact of these genetic factors among tumor subtype. Third, there are considerable difference between ESCC and GC, including environment and patient characteristics, which may have introduced potential bias on our results. Therefore, our results were preliminary and should be further evaluated in future studies.
In summary, we evaluated the genetic association between EC and GC, and found shared genetic susceptibility between ESCC and GC. In the future, more studies with larger sample sizes and multiple populations are needed to help detect the relationship between the genetic basis of EC and GC.
References
Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359–386.
Van Cutsem E, Sagaert X, Topal B, Haustermans K, Prenen H. Gastric cancer. Lancet. 2016;388:2654–64.
Domper Arnal MJ, Ferrandez Arenas A, Lanas Arbeloa A. Esophageal cancer: Risk factors, screening and endoscopic treatment in Western and Eastern countries. World J Gastroenterol. 2015;21:7933–43.
Pennathur A, Gibson MK, Jobe BA, Luketich JD. Oesophageal carcinoma. Lancet. 2013;381:400–12.
Vaughan TL, Davis S, Kristal A, Thomas DB. Obesity, alcohol, and tobacco as risk factors for cancers of the esophagus and gastric cardia: adenocarcinoma versus squamous cell carcinoma. Cancer Epidemiol Biomarkers Prev. 1995;4:85–92.
Morita M, Kumashiro R, Kubo N, Nakashima Y, Yoshida R, Yoshinaga K, et al. Alcohol drinking, cigarette smoking, and the development of squamous cell carcinoma of the esophagus: epidemiology, clinical findings, and prevention. Int J Clin Oncol. 2010;15:126–34.
Brown LM, Silverman DT, Pottern LM, Schoenberg JB, Greenberg RS, Swanson GM, et al. Adenocarcinoma of the esophagus and esophagogastric junction in white men in the United States: alcohol, tobacco, and socioeconomic factors. Cancer Causes Control. 1994;5:333–40.
Lee CH, Wu DC, Lee JM, Wu IC, Goan YG, Kao EL, et al. Carcinogenetic impact of alcohol intake on squamous cell carcinoma risk of the oesophagus in relation to tobacco smoking. Eur J Cancer. 2007;43:1188–99.
Runge TM, Abrams JA, Shaheen NJ. Epidemiology of Barrett’s esophagus and esophageal adenocarcinoma. Gastroenterol Clin North Am. 2015;44:203–31.
Abnet CC, Freedman ND, Hu N, Wang Z, Yu K, Shu XO, et al. A shared susceptibility locus in PLCE1 at 10q23 for gastric adenocarcinoma and esophageal squamous cell carcinoma. Nat Genet. 2010;42:764–7.
Gharahkhani P, Fitzgerald RC, Vaughan TL, Palles C, Gockel I, Tomlinson I, et al. Genome-wide association studies in oesophageal adenocarcinoma and Barrett’s oesophagus: a large-scale meta-analysis. Lancet Oncol. 2016;17:1363–73.
Shi Y, Hu Z, Wu C, Dai J, Li H, Dong J, et al. A genome-wide association study identifies new susceptibility loci for non-cardia gastric cancer at 3q13.31 and 5p13.1. Nat Genet. 2011;43:1215–8.
Cui R, Kamatani Y, Takahashi A, Usami M, Hosono N, Kawaguchi T, et al. Functional variants in ADH1B and ALDH2 coupled with alcohol and smoking synergistically enhance esophageal cancer risk. Gastroenterology. 2009;137:1768–75.
Wang LD, Zhou FY, Li XM, Sun LD, Song X, Jin Y, et al. Genome-wide association study of esophageal squamous cell carcinoma in Chinese subjects identifies susceptibility loci at PLCE1 and C20orf54. Nat Genet. 2010;42:759–63.
Wu C, Hu Z, He Z, Jia W, Wang F, Zhou Y, et al. Genome-wide association study identifies three new susceptibility loci for esophageal squamous-cell carcinoma in Chinese populations. Nat Genet. 2011;43:679–84.
Abnet CC, Wang Z, Song X, Hu N, Zhou FY, Freedman ND, et al. Genotypic variants at 2q33 and risk of esophageal squamous cell carcinoma in China: a meta-analysis of genome-wide association studies. Hum Mol Genet. 2012;21:2132–41.
Wu C, Kraft P, Zhai K, Chang J, Wang Z, Li Y, et al. Genome-wide association analyses of esophageal squamous cell carcinoma in Chinese identify multiple susceptibility loci and gene-environment interactions. Nat Genet. 2012;44:1090–7.
Wu C, Wang Z, Song X, Feng XS, Abnet CC, He J, et al. Joint analysis of three genome-wide association studies of esophageal squamous cell carcinoma in Chinese populations. Nat Genet. 2014;46:1001–6.
McKay JD, Truong T, Gaborieau V, Chabrier A, Chuang SC, Byrnes G, et al. A genome-wide association study of upper aerodigestive tract cancers conducted within the INHANCE consortium. PLoS Genet. 2011;7:e1001333.
Delaneau O, Marchini J, Zagury JF. A linear complexity phasing method for thousands of genomes. Nat Methods. 2011;9:179–81.
Howie BN, Donnelly P, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 2009;5:e1000529.
Wing MR, Bourdon DM, Harden TK. PLC-epsilon: a shared effector protein in Ras-, Rho-, and G alpha beta gamma-mediated signaling. Mol Interv. 2003;3:273–80.
Harden TK, Sondek J. Regulation of phospholipase C isozymes by ras superfamily GTPases. Annu Rev Pharmacol Toxicol. 2006;46:355–79.
Hu H, Yang J, Sun Y, Yang Y, Qian J, Jin L, et al. Putatively functional PLCE1 variants and susceptibility to esophageal squamous cell carcinoma (ESCC): a case-control study in eastern Chinese populations. Ann Surg Oncol. 2012;19:2403–10.
Zhang H, Jin G, Li H, Ren C, Ding Y, Zhang Q, et al. Genetic variants at 1q22 and 10q23 reproducibly associated with gastric cancer susceptibility in a Chinese population. Carcinogenesis. 2011;32:848–52.
Palmer AJ, Lochhead P, Hold GL, Rabkin CS, Chow WH, Lissowska J, et al. Genetic variation in C20orf54, PLCE1 and MUC1 and the risk of upper gastrointestinal cancers in Caucasian populations. Eur J Cancer Prev. 2012;21:541–4.
Haddad SA, Ruiz-Narvaez EA, Haiman CA, Sucheston-Campbell LE, Bensen JT, Zhu Q, et al. An exome-wide analysis of low frequency and rare variants in relation to risk of breast cancer in African American Women: the AMBER Consortium. Carcinogenesis. 2016;37:870–7.
Natrajan R, Mackay A, Lambros MB, Weigelt B, Wilkerson PM, Manie E, et al. A whole-genome massively parallel sequencing analysis of BRCA1 mutant oestrogen receptor-negative and -positive breast cancers. J Pathol. 2012;227:29–41.
Pullamsetti SS, Banat GA, Schmall A, Szibor M, Pomagruk D, Hanze J, et al. Phosphodiesterase-4 promotes proliferation and angiogenesis of lung cancer by crosstalk with HIF. Oncogene. 2013;32:1121–34.
Rahrmann EP, Collier LS, Knutson TP, Doyal ME, Kuslak SL, Green LE, et al. Identification of PDE4D as a proliferation promoting factor in prostate cancer using a Sleeping Beauty transposon-based somatic mutagenesis screen. Cancer Res. 2009;69:4388–97.
Powers GL, Hammer KD, Domenech M, Frantskevich K, Malinowski RL, Bushman W, et al. Phosphodiesterase 4D inhibitors limit prostate cancer growth potential. Mol Cancer Res. 2015;13:149–60.
Bottcher R, Dulla K, van Strijp D, Dits N, Verhoef EI, Baillie GS, et al. Human PDE4D isoform composition is deregulated in primary prostate cancer and indicative for disease progression and development of distant metastases. Oncotarget. 2016;7:70669–84.
Henderson DJ, Byrne A, Dulla K, Jenster G, Hoffmann R, Baillie GS, et al. The cAMP phosphodiesterase-4D7 (PDE4D7) is downregulated in androgen-independent prostate cancer cells and mediates proliferation by compartmentalising cAMP at the plasma membrane of VCaP prostate cancer cells. Br J Cancer. 2014;110:1278–87.
Chen, ZH, Xian, JF & Luo, LP Analysis of ADH1B Arg47His, ALDH2 Glu487Lys, and CYP4502E1 polymorphisms in gastric cancer risk and interaction with environmental factors. Genet Mol Res. 15 (2016).
Duell EJ, Sala N, Travier N, Munoz X, Boutron-Ruault MC, Clavel-Chapelon F, et al. Genetic variation in alcohol dehydrogenase (ADH1A, ADH1B, ADH1C, ADH7) and aldehyde dehydrogenase (ALDH2), alcohol consumption and gastric cancer risk in the European Prospective Investigation into Cancer and Nutrition (EPIC) cohort. Carcinogenesis. 2012;33:361–7.
Tsai ST, Wong TY, Ou CY, Fang SY, Chen KC, Hsiao JR, et al. The interplay between alcohol consumption, oral hygiene, ALDH2 and ADH1B in the risk of head and neck cancer. Int J Cancer. 2014;135:2424–36.
Guo XF, Wang J, Yu SJ, Song J, Ji MY, Zhang JX, et al. Meta-analysis of the ADH1B and ALDH2 polymorphisms and the risk of colorectal cancer in East Asians. Intern Med. 2013;52:2693–9.
Liu, P, Zhao, HR, Li, F, Zhang, L, Zhang, H, Wang, WR et al. Correlations of ALDH2 rs671 and C12orf30 rs4767364 polymorphisms with increased risk and prognosis of esophageal squamous cell carcinoma in the Kazak and Han populations in Xinjiang province. J Cin Lab Anal. 32 (2018).
Gao Y, He Y, Xu J, Xu L, Du J, Zhu C, et al. Genetic variants at 4q21, 4q23 and 12q24 are associated with esophageal squamous cell carcinoma risk in a Chinese population. Hum Genet. 2013;132:649–56.
Yu, C, Guo, Y, Bian, Z, Yang, L, Millwood, IY, Walters, RG et al. Association of low-activity ALDH2 and alcohol consumption with risk of esophageal cancer in Chinese adults: a population-based cohort study. Int J Cancer. (2018).
Hidaka A, Sasazuki S, Matsuo K, Ito H, Sawada N, Shimazu T, et al. Genetic polymorphisms of ADH1B, ADH1C and ALDH2, alcohol consumption, and the risk of gastric cancer: the Japan Public Health Center-based prospective study. Carcinogenesis. 2015;36:223–31.
Yang S, Lee J, Choi IJ, Kim YW, Ryu KW, Sung J, et al. Effects of alcohol consumption, ALDH2rs671 polymorphism, and Helicobacter pylori infection on the gastric cancer risk in a Korean population. Oncotarget. 2017;8:6630–41.
Zhang LQ, Song X, Zhao XK, Huang J, Zhang P, Wang LW, et al. Association of genotypes of rs671 withinALDH2 with risk for gastric cardia adenocarcinoma in the Chinese Han population in high- and low-incidence areas. Cancer Biol Med. 2017;14:60–65.
Shin CM, Kim N, Cho SI, Kim JS, Jung HC, Song IS. Association between alcohol intake and risk for gastric cancer with regard to ALDH2 genotype in the Korean population. Int J Epidemiol. 2011;40:1047–55.
Testino G, Leone S, Patussi V, Scafato E. [Alcohol, cardiovascular prevention and cancer]. Recent Prog Med. 2014;105:144–6.
Haas SL, Ye W, Lohr JM. Alcohol consumption and digestive tract cancer. Curr Opin Clin Nutr Metab Care. 2012;15:457–67.
Acknowledgements
We thank all the participants of the Nanjing/Beijing and the National Cancer Institute gastric cancer studies. This study was supported by grants from the National key research and development program of China (grant no. 2016YFC1302703); and National Natural Science Foundation of China (81521004, 81422042, 81872702).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Human rights statement and informed consent
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1964 and later versions. Informed consent to be included in the study, or the equivalent, was obtained from all patients.
Additional information
Authors share co-first authorship: Linhua Yao, Fei Yu, Yingying Mao
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Yao, L., Yu, F., Mao, Y. et al. Gastric cancer may share genetic predisposition with esophageal squamous cell carcinoma in Chinese populations. J Hum Genet 63, 1159–1168 (2018). https://doi.org/10.1038/s10038-018-0501-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s10038-018-0501-4
