
Abstract
Intracranial vertebral–basilar artery dissection (IVAD) is an arterial disorder leading to life-threatening consequences. Genetic factors are known to be causative to certain syndromic forms of IVAD. However, systematic study of the molecular basis of sporadic and isolated IVAD is lacking. To identify genetic variants contributing to the etiology of IVAD, we enrolled a cohort of 44 unrelated cases with a clinical diagnosis of isolated IVAD and performed whole-exome sequencing (WES) for all the participants; a trio exome sequencing approach was used when samples from both parents were available. Four previously reported disease-causing heterozygous variants (three in COL3A1 and one in FBN1) and seven novel heterozygous variants in IVAD-related genes were identified. In addition, six variants in novel IVAD genes including two de novo heterozygous nonsynonymous variants (each in VPS52 and CDK18), two stop-gain variants (each in MYH9 and LYL1), and two heterozygous biallelic variants in TNXB were considered to be possibly contributing to the phenotype, with unknown significance according to the existing knowledge. A significantly higher mutational rate of IVAD candidate genes was observed in patients versus our in-house controls (P = 0.002) (DISCO study, http://www.discostudy.org/, n = 2248). Our study provided a mutational landscape for patients with isolated IVAD.
Similar content being viewed by others
Introduction
Intracranial vertebral–basilar artery dissection (IVAD) is a pathological condition of the artery caused by the diversion of circulating blood into the weakened vessel wall, with an incidence of 1–1.5 per 100,000 individuals every year [1]. IVAD can lead to life-threatening consequences if diagnoses are delayed, accompanied by symptoms such as hemorrhage, mass effect, and stroke [1,2,3], with a high mortality ranging from 19 to 83% [3].
While IVAD is usually nonsyndromic, it can be present in congenital syndromes featuring connective tissue disorders, such as vascular Ehlers–Danlos syndrome [4], Marfan syndrome [5], and Loeys–Dietz syndrome [6]. Considering the occurrence of familial aggregation and the high prevalence of histological abnormalities of connective tissues in patients with sporadic arterial dissections, genetic factors were assumed to also underlie nonsyndomic vertebral artery dissection [5, 7]. Genome-wide association studies (GWASs) have been utilized extensively to locate genomic markers related to increased risk of IVAD. An increased prevalence of the c.677C>T genotype of methylenetetrahydrofolate reductase (MTHFR) was reported in cervical artery dissection (CeAD) patients [8]. The E496K protein change in intercellular adhesion molecule-1 (ICAM-1) was enriched in patients with spontaneous CeAD [9], suggesting that proinflammatory factors might be involved in the etiology of CeAD. Recently, a common allele rs9349379[G] of PHACTR1 was found by GWAS to be associated with a reduced risk of both vertebral and carotid artery dissection [10].
The application of next-generation sequencing (NGS) to identify pathogenic or risk variants in patient cohorts with IVAD-associated diseases has also been reported [11, 12]. Using a panel of 16 genes, a missense variant c.953C>G in COL3A1 was identified in a CeAD pedigree [11]. Exome sequencing of 15 CAD and 3 VAD patients identified significantly higher rate of nonbenign variants in FBN1, TGFBR2, COL3A1, and COL4A1 [12]. The approaches of NGS have so far focused on genes related to connective tissue disorders due to their known association with artery dissection.
In order to decipher the molecular basis of IVAD at an exome level, we herein performed a whole-exome sequencing (WES) study interrogating a cohort of patients with IVAD. In addition to identifying variants in known arterial dissection-related genes, we also identified potentially contributory variants in novel IVAD genes.
Methods
Cohort collection
We consecutively enrolled cases diagnosed with IVAD from January 2016 to December 2016 at Beijing Tiantan Hospital, China. Digital subtraction angiography, computed tomographic angiography, magnetic resonance imaging, and magnetic resonance angiography were used for radiological assessment. The inclusion and exclusion criteria and detailed phenotypic data are provided in the Supplementary material. This study was approved by the ethics committee of Beijing Tiantan Hospital. Informed consent was obtained from each patient or their parents.
Whole-exome sequencing
WES was performed on peripheral blood for all the participants. In brief, DNA samples were prepared into Illumina paired-end libraries and underwent whole-exome capture with the SureSelect Human All Exon V6+UTR r2 core design (91 Mb, Agilent), followed by sequencing on the Illumina HiSeq 4000 platform (Illumina, San Diego, CA, USA). In-house-developed Peking Union Medical college hospital Pipeline (PUMP) and variant interpretation methods are provided in the Supplementary Methods.
WES data interpretation
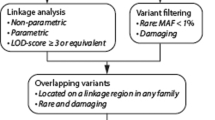
The detailed variant calling and annotation methods are provided in the Supplementary Material. After filtering, variants in 25 genes related with artery/aortic dissection according to the Human Gene Mutation Database (HGMD) and the Online Mendelian Inheritance in Man (OMIM) (Table S1) were analyzed. Reported pathogenic variants in IVAD candidate genes were classified as ‘disease-causing variants’. Novel variants in IVAD genes are classified as ‘susceptibility variants’. Apart from variants in reported genes, additional variants were selected based on their pathogenicity and inheritance patterns (Fig. 1). Firstly, truncating variants in genes predicted to be intolerant to loss-of-function changes according to the Exome Aggregation Consortium (ExAC) database (probability of loss-of-function intolerance (pLI) score ≥0.9, or having no truncating variants) were identified as susceptibility variants. De novo and biallelic variants (compound heterozygous and homozygous variants) identified by trio analysis were also added to the category of susceptibility variants.

An overview of the variant prioritization workflow. MAF minor allele frequency, pLI probability of loss-of-function intolerance, novel absent from public databases (ExAC, 1000 genome, ESP5400)
In-house control database
In-house control database consisted of 2248 individuals without apparent vascular deformity from Peking Union Medical College Hospital as part of the Deciphering Disorders Involving Scoliosis and COmorbidities (DISCO) study (http://www.discostudy.org/). WES was performed on peripheral blood using the same protocol.
Rare variant burden analyses
To alleviate confounding factors attributable to differential sequencing coverage, we conducted harmonization analyses between case and control exomes. An individual RefSeq coding sequence site was excluded from the analysis if the absolute difference in percentages of cases compared to controls with adequate coverage of the site differed by greater than 8.2%. This site-based pruning resulted in exclusion of 4.8% of the RefSeq coding sequence sites.
After site-based pruning analyses, rare nonsynonymous variants (missense, nonsense, frameshift, in-frame indel, and canonical splicing site) were extracted to capture the category of genetic variation that meets specific criteria designed to enrich for pathogenic variants. To identify rare variants, we use internal (combined case and control population) and external (ExAC and gnomAD) sequence data to filter for variants with a minor allele frequency (MAF) < 0.01.
Rare variants were analyzed using SNP-set (Sequence) Kernel Association Test-Optimized (SKAT-O) test to determine the association of mutational burden in the 25 genes. To correct for multiple testing, Benjamini–Hochberg method was applied.
Results
Cohort information
We enrolled a total of 8 cases with unaffected parents (trios) and 36 singleton cases of Chinese Han ethnicity, consisting of 35 males and 9 females, with a mean age of diagnosis of 42.1 ± 17.7 years. Seven children (younger than 14 years) accounted for 16% of all the patients. Clinical classification was performed according to Zhang et al. [13]. Detailed demographic information is presented in Table 1.
Whole-exome sequencing identified 17 candidate variants
The detailed WES quality parameter is shown in Supplementary Materials (Table S2). After manual review and prioritization, we identified 17 variants possibly contributing to the IVAD phenotype of 15 patients, including 11 variants in known IVAD-related genes and 6 variants in possible novel disease genes (Table 2). All candidate variants were confirmed by Sanger sequencing. We classified these variants into two tiers (4 for disease-causing variants and 13 for susceptibility variants), in which disease-causing variants demonstrate reported pathogenicity (Fig. 1), while the susceptibility variants required additional evidence to establish their contribution to the disease.
Mutational spectrums revealed for known IVAD candidate genes
Because the genetic factors underlying nonsyndromic IVAD have only recently begun to be understood, we first examined the 25 genes known to cause connective tissue disorders manifesting artery or aortic dissections according to the HGMD and OMIM databases (Table S1).
Previously reported disease-causing variants were found in two genes, COL3A1 (n = 3) and FBN1 (n = 1) (Table 2). Two reported heterozygous variants in COL3A1, c.3133G>A (p.Ala1045Thr) and c.1815+5G>A, were observed in patient IVAD49, with unknown chromosomal phases due to a lack of parental samples. Interestingly, the c.1815+5G>A variant has been reported in a patient affected with vascular Ehlers–Danlos syndrome [14] and a 67-year-old female with thoracic aortic aneurysm dissection and hyperlipidemia [15]. Although the patient with Ehlers–Danlos syndrome showed no sign of artery disease, histological examination demonstrated abnormality in α1(III) collagen chains, and reverse transcription-PCR confirmed the skipping of exon [14], supporting the pathogenicity of the c.1815+5G>A variant. The c.3133G>A (p.Ala1045Thr) variant has been observed once in a cohort of aortic aneurysm and dissection patients [15]. The third known heterozygous variant in COL3A1, c.1258G>A (p.Gly420Ser), of unknown inheritance was identified in patient IVAD61. The same allelic change, which is absent in the ExAC database, was reported in a 41-year-old woman with dissecting of mesenteric artery and right vertebral artery, with the left vertebral artery showing intramural hemorrhage and patchy fibromuscular disorganization [16]. Consistent with the reported phenotypes, IVAD61 is a 54-year-old woman who had dissecting aneurysm of both left and right vertebral arteries. In an attempt to explore the potential genotype–phenotype correlation of COL3A1, we compared the clinical features of IVAD49 and IVAD61 (Table 3). They both had artery dissection at the section of the vertebral artery (35/44 in the cohort), and neither of them was affected with hypertension (26/44 in the cohort). Due to the limited sample size and viable expressivity, we were unable to characterize the clinical features specific to COL3A1-related IVAD.
In addition, we identified one reported heterozygous FBN1 variant c.3455C>T (p.Ala1152Val) of unknown inheritance from patient IVAD51. The variant was described in a patient with Marfan syndrome manifesting cardiovascular problems and dural ectasia [17].
In an effort to extend the mutational spectrums and disease contribution for the IVAD-related genes mentioned above, we incorporated our in-house control of 2248 exomes without apparent vascular deformities, and performed a mutational burden test upon the number of qualified variants in these 25 genes defined as rare (MAF < 0.01) nonsense/frameshift/in-frame indel/nonsynonymous variants with high-quality reads.
As a result, we identified 44 qualified variants from our cohort and 1572 from the in-house controls. The average number of qualified variants in our IVAD patients (1.0) is significantly higher than that in the in-house controls (0.7) (P = 0.002, SKAT-O) (Table S3), suggesting that the mutational burden of IVAD-related genes likely contributed to the disease etiology.
On the variant level, we analyzed variants in IVAD genes that have not been observed in either public or in-house control populations, also taking into account the disease-causing model (dominant or recessive) of these genes. We identified six novel heterozygous variants in FBN1, COL5A2, NOTCH1, and PDCD10, and one novel hemizygous variant in FLNA from nine cases (Table 2). These novel variants are currently considered as variants of unknown significance and are consequently grouped into susceptibility variants in this study.
Potential novel IVAD-related disease genes
In order to uncover novel disease genes potentially causing IVAD, we focused on new candidate genes previously not associated with IVAD-related syndromes, but linked to relevant biological processes. We identified two de novo missense variants, two stop-gain variants, and two compound heterozygous variants from five genes (Table 2).
A heterozygous de novo variant c.1492C>T (p.Arg498Cys) was identified in VPS52 from a trio family (IVAD5 and parents). The patient was a 5-year-old female with a dissecting aneurysm of the left vertebral artery. She was admitted for dizziness and severe headache. Intracranial angiography showed a dolichoectatic dissecting aneurysm of the left vertebral artery (Fig. 2a). VPS52 encodes a highly conserved protein forming part of the GARP (Golgi associated retrograde protein) complex [18]. Epiblast lineage-specific null mutants showed abnormal vasculogenesis in mouse yolk sac, resulting in blood congestion [19]. The missense variant was absent from public databases and had substantial damaging predictions by bioinformatics tools (gerp++ = 5.19, combined annotation dependent depletion (CADD) = 22.7, sorting intolerant from tolerant (SIFT) = 0.01, and polymorphism phenotyping v2 (Polyphen2) = 1). The substitution is located in a highly conserved segment on the outer surface of VPS52 protein, and could possibly affect its interaction with other components of the GARP complex or induce a gain-of-function effect (Fig. 2a). The three-dimensional structure of the VPS52 protein provided further evidence of pathogenicity, as the long side chain of Arginine528 was replaced by a short, sulfur-containing side chain of cysteine (Fig. 2a).

Clinical and genetic characteristics of patients carrying variants in novel IVAD-related genes. a Left, Sanger validation of the VPS52 variant in IVAD5. Middle-left, left vertebral angiogram (lateral view) reveals a dolichoectatic dissecting aneurysm (arrow) of V4 segment of the left vertebral artery. Axial high-resolution T2WI (middle-right) shows a giant dissecting aneurysm with pseudolumen (arrow) of the left vertebral artery and its serious mass effect. Right, protein structure predicted by I-TASSER shows replacement of the long side chain of Arginine528 by a short, sulfur-containing side chain of cysteine. Surface structure modeling shows that the Arg498 was located at the surface of the protein. b Left, Sanger validation of the CDK18 variant in IVAD23. Middle-left, left vertebral angiogram (frontal view) reveals a junctional dissecting aneurysm (arrow) of vertebral artery V4 segment and basilar artery in IVAD23. Axial high-resolution T2WI (middle-right) shows left vertebral artery harboring a giant dissection (arrow) with thrombosis inside the lumen. Right, the p.Lys142Asn variant alters a conserved residue. c Left, Sanger validation of the LYL1 variant in IVAD53. Middle-left, left vertebral angiogram (frontal view) illustrates a dissecting aneurysm (arrow) of V4 segment of the left vertebral artery. Middle-right, Axial high-resolution T2WI displays left vertebral artery dissection (arrow) with pseudolumen. Right, the NM_005583.4: c.748C>T (p.Gln250Ter) variant creates a premature stop codon close to the C terminus of the LYL1 protein. d Left, Sanger validation of the MYH9 variant in IVAD73. Middle-left, left vertebral angiogram (lateral view) reveals a dissecting aneurysm of the V4 segment of the left vertebral artery. Middle-right, Axial high-resolution T2WI confirms left vertebral artery dissection (arrow). Right, the NM_002473.4:c.487C>T(p.Gln163Ter) variant (in black) generates a premature stop codon at the beginning of the motor domain. Three nonsense variants (in gray) previously reported to cause MYH9-related disorder according to HGMD are located near the C terminal
A heterozygous de novo variant in the CDK18 genec.426G>C (p.Lys142Asn) was identified from another trio family (IVAD23 and parents) (Fig. 2b). This variant is predicted to be substantially deleterious (gerp++ = 5.14, CADD = 27.9, SIFT = 0.04, and Polyphen2 = 0.996). IVAD23 was an 8-year-old female who presented intermittent headache for 3 weeks. Left vertebral angiogram revealed a large dissecting aneurysm (20 × 25 mm) of the initial segment of her basilar artery (Fig. 2b). CDK18 is highly expressed in the human brain and may affect the regulation of cell cycle as a cyclin-dependent kinase [20], thus possibly disrupting the normal growth of brain vessels. CDK18 has not been previously associated with arterial dissection. Therefore, this finding may represent a novel disease association for CDK18.
Truncating variants were identified in two genes that are potentially associated with IVAD among the singletons. A heterozygous stop-gain variant of c.748C>T (p.Gln250Ter) was identified in LYL1 from IVAD53, a 51-year-old female with a dissecting aneurysm of the left vertebral artery. The patient had a persistent headache that lasted for 1 month. Intracranial angiography revealed classic dissecting aneurysm accompanied with proximal stenosis of the left vertebral artery (Fig. 2c). LYL1−/− mice showed accelerated tumor growth and angiogenesis. Moreover, enhanced angiogenesis is accompanied by a strong reduction of blood vessel coverage by mural cells, resulting in leaky blood vessels [21]. Heterozygous loss-of-function variants in LYL1 have not been reported from humans in the ExAC database. The premature stop codon is located near the 3′ end of the coding sequence (a total length of 280 amino acid) on the last exon (Fig. 2c), possibly resulting in escaping of the nonsense-mediated mRNA decay (NMD) and inducing abnormal folding of the protein. Taken together, our observations implicate that LYL1 deficiency could contribute to IVAD in humans.
A heterozygous stop-gain variant c.487C>T (p.Gln163Ter) was found in MYH9 in a singleton IVAD73, a 61-year-old male with a dissecting aneurysm of the left vertebral artery. The patient had persistent headaches for 3 years. Intracranial angiography showed dolichoectatic dissecting aneurysm of the left vertebral artery (Fig. 2d). MYH9 is required for macrophage human antigen R (HuR) translocation and vascular endothelial growth factor-A (VEGF-A) messenger RNA (mRNA) stabilization, a potential contributor to arteriogenesis [22]. Defects in MYH9 have been demonstrated to cause a spectrum of clinical conditions called MYH9-related disorders (MYH9-RD). Phenotypes of MYH9-RD include macrothrombocytopenia, leukocyte inclusions, and hearing loss [23], and the pathogenic mechanism of MYH9-RD is believed to be associated with a dominant-negative effect [24]. Three nonsense variants reported to cause MYH9-RD according to HGMD are located near the C terminal of the protein and might escape NMD (Fig. 2d). The variant observed in patient IVAD73 generates a premature stop codon in the exon 3 of MYH9 (Fig. 2d). The consequence of haploinsufficiency of MYH9 has not been studied. However, MYH9 is predicted to be intolerant to loss-of-function alterations, based on the analysis from the ExAC database (pLI = 1). Therefore, the potential loss-of-function variant in MYH9 observed in the IVAD patient may represent a distinct disease mechanism causing a novel IVAD-related disorder that is different from MYH9-RDs.
Another rare heterozygous missense variant c.1178G>A (p.Arg393His) in MYH9 was identified in IVAD47 (Table S4), with ExAC allele frequency of 0.000074 and robust damaging prediction by bioinformatics tools (gerp++ = 4.96, CADD = 35, SIFT = 0, Polyphen2 = 1.0). IVAD47 had dissecting aneurysm of left vertebral artery.
Biallelic variants of unknown significance were identified in TNXB in our patient cohort (Table 2). Compound heterozygous TNXB variants c.7826–4C>T and c.7802C>T (p.Pro2601Leu) were found in IVAD82, and other possible biallelic TNXB variants were observed in two singletons (Table S4). TNXB has been reported to relate with classical-like Ehlers–Danlos syndrome [4, 25], but not currently known to cause a vascular subtype of Ehlers–Danlos syndrome. Therefore, our findings may represent a phenotypic expansion for this gene.
Discussion
In this study, we used a systematic approach to investigate the potential genetic mechanism underlying IVAD. We identified known and novel variants in known IVAD-related genes and variants in potential novel genes.
Fewer than 2% of all IVAD cases occur as a complication of known monogenic connective syndrome disorders, mostly vascular Ehlers–Danlos syndrome, according to the previous large study series [26]. However, rare deleterious mutations in COL3A1 have been linked to nonsyndromic artery dissections, with no manifestation of vascular Ehlers–Danlos syndrome [16, 27], suggesting genetic pleiotropy for COL3A1. Our findings of pathogenic variants in COL3A1 replicated the previous studies and confirmed the causal role of these mutations in nonsyndromic IVAD [16].
Common variants have been found to be associated with increased or decreased risk of VAD or CAD [10], but have not fully explained the etiology or provided guidance for clinical management or genetic counseling [28]. Based on the results from this study and previous studies, the etiology of isolated IVAD is unlikely to be fully explained by a monogenetic model for a fraction of patients, even with the concepts of incomplete penetrance and variable expressivity [29]. We identified a significant higher mutational rate of IVAD-associated candidate genes by performing a burden test upon 44 IVAD patients and 2248 in-house controls. Our results suggest that rare variants in 25 IVAD-associated genes could contribute to the genetic background leading to IVAD, although most of them are not sufficient to cause the disease independently.
Most previous efforts to reveal the genetic basis of IVAD examined known connective tissue-related genes using subjects with a family history from large pedigrees [11, 12]. In contrast, our study design focused on unrelated sporadic IVAD cases, which allows us to investigate the genetic etiology of a specific subtype of artery dissection in a more realistic and practical clinical point of view. Moreover, our utilization of exome sequencing enabled the possibility of discovering novel disease genes potentially related to IVAD. We identified four genes that harbor potential contributory variants, which have biological relationship with IVAD.
The de novo origin of VPS52 and CDK18 variants are consistent with the sporadic occurrence of IVAD in two families. However, further investigation is in need to test the function of these missense variants before their causal role could be confirmed. In contrast, truncating variants in LYL1 and MYH9 are most likely to be pathogenic, supported by the prediction of loss-of-function intolerance score from ExAC database. Nevertheless, the relationship between haploinsufficiency of these two genes and IVAD phenotype still demands further studies.
Compound heterozygous and potential compound heterozygous TNXB variants were identified in three patients, consistent with the inheritance mode in which TNXB causes classical-like Ehlers–Danlos syndrome [4, 25]. Aside from the common Ehlers–Danlos syndrome phenotypes such as joint hypermobility and skin hyperextensibility, vesicoureteral reflux [30] has been linked to TNXB mutations, suggesting its phenotypic pleiotropy. Although TNXB has not been associated with vascular deformities, TNX proteins are crucial for the activation of transforming growth factor-β (TGF-β) and subsequent epithelial-to-mesenchymal transition [31], implicating a potential role in maintaining vascular integrity.
A small sample size represents the main limitation of our study. Replication of the burden test signal and novel candidate genes in future larger cohorts is warranted. Functional validations of detected variants should also be conducted in further studies.
To summarize, we performed WES on a sporadic IVAD cohort composed of 8 trios and 36 singletons. Our findings of previously reported variants strengthened the pathogenicity of those variants. Mutational burden test on IVAD candidate genes supported their contribution to the genetic background of IVAD. We also identified novel susceptible variants in known IVAD candidate genes (FBN1, COL5A2, NOTCH1, PDCD10, and FLNA) and in novel candidate genes (VPS52, CDK18, LYL1, and MYH9).
References
Balik V, Yamada Y, Talari S, Kei Y, Sano H, Suyama D, et al. State-of-art in surgical treatment of dissecting posterior circulation intracranial aneurysms. Neurosurg Rev. 2016;41:31–45.
Kaufmann TJ, Huston J 3rd, Mandrekar JN, Schleck CD, Thielen KR, Kallmes DF. Complications of diagnostic cerebral angiography: evaluation of 19,826 consecutive patients. Radiology. 2007;243:812–9.
Ramgren B, Cronqvist M, Romner B, Brandt L, Holtas S, Larsson EM. Vertebrobasilar dissection with subarachnoid hemorrhage: a retrospective study of 29 patients. Neuroradiology. 2005;47:97–104.
Malfait F, Francomano C, Byers P, Belmont J, Berglund B, Black J, et al. The 2017 international classification of the Ehlers-Danlos syndromes. Am J Med Genet C Semin Med Genet. 2017;175:8–26.
Giossi A, Ritelli M, Costa P, Morotti A, Poli L, Del Zotto E, et al. Connective tissue anomalies in patients with spontaneous cervical artery dissection. Neurology. 2014;83:2032–7.
Fujita D, Takeda N, Morita H, Kato M, Nishimura H, Inuzuka R, et al. A novel mutation of TGFBR2 causing Loeys-Dietz syndrome complicated with pregnancy-related fatal cervical arterial dissections. Int J Cardiol. 2015;201:288–90.
Grond-Ginsbach C, Debette S, Pezzini A. Genetic approaches in the study of risk factors for cervical artery dissection. Front Neurol Neurosci. 2005;20:30–43.
Pezzini A, Del Zotto E, Archetti S, Negrini R, Bani P, Albertini A, et al. Plasma homocysteine concentration, C677T MTHFR genotype, and 844ins68bp CBS genotype in young adults with spontaneous cervical artery dissection and atherothrombotic stroke. Stroke. 2002;33:664–9.
Longoni M, Grond-Ginsbach C, Grau AJ, Genius J, Debette S, Schwaninger M, et al. The ICAM-1 E469K gene polymorphism is a risk factor for spontaneous cervical artery dissection. Neurology. 2006;66:1273–5.
Debette S, Kamatani Y, Metso TM, Kloss M, Chauhan G, Engelter ST, et al. Common variation in PHACTR1 is associated with susceptibility to cervical artery dissection. Nat Genet. 2015;47:78–83.
Makrygiannis G, Loeys B, Defraigne JO, Sakalihasan N. Cervical artery dissections and type A aortic dissection in a family with a novel missense COL3A1 mutation of vascular type Ehlers-Danlos syndrome. Eur J Med Genet. 2015;58:634–6.
Grond-Ginsbach C, Brandt T, Kloss M, Aksay SS, Lyrer P, Traenka C, et al. Next generation sequencing analysis of patients with familial cervical artery dissection. Eur Stroke J. 2017;2:137–43.
Zhang Y, Tian Z, Sui B, Wang Y, Liu J, Li M, et al. Endovascular treatment of spontaneous intracranial fusiform and dissecting aneurysms: outcomes related to imaging classification of 309 cases. World Neurosurg. 2017;98:444–55.
Lee B, Vitale E, Superti-Furga A, Steinmann B, Ramirez F. G to T transversion at position +5 of a splice donor site causes skipping of the preceding exon in the type III procollagen transcripts of a patient with Ehlers-Danlos syndrome type IV. J Biol Chem. 1991;266:5256–9.
Sakai H, Suzuki S, Mizuguchi T, Imoto K, Yamashita Y, Doi H, et al. Rapid detection of gene mutations responsible for non-syndromic aortic aneurysm and dissection using two different methods: resequencing microarray technology and next-generation sequencing. Hum Genet. 2012;131:591–9.
Pickup MJ, Pollanen MS. Traumatic subarachnoid hemorrhage and the COL3A1 gene: emergence of a potential causal link. Forensic Sci Med Pathol. 2011;7:192–7.
Hung CC, Lin SY, Lee CN, Cheng HY, Lin SP, Chen MR, et al. Mutation spectrum of the fibrillin-1 (FBN1) gene in Taiwanese patients with Marfan syndrome. Ann Hum Genet. 2009;73:559–67.
Conibear E, Stevens TH. Vps52p, Vps53p, and Vps54p form a novel multisubunit complex required for protein sorting at the yeast late Golgi. Mol Biol Cell. 2000;11:305–23.
Sugimoto M, Kondo M, Hirose M, Suzuki M, Mekada K, Abe T, et al. Molecular identification of t(w5): Vps52 promotes pluripotential cell differentiation through cell-cell interactions. Cell Rep. 2012;2:1363–74.
Herskovits AZ, Davies P. Cloning and expression analysis of two novel PCTAIRE 3 transcripts from human brain. Gene. 2004;328:59–67.
Pirot N, Deleuze V, El-Hajj R, Dohet C, Sablitzky F, Couttet P, et al. LYL1 activity is required for the maturation of newly formed blood vessels in adulthood. Blood. 2010;115:5270–9.
Morrison AR, Yarovinsky TO, Young BD, Moraes F, Ross TD, Ceneri N, et al. Chemokine-coupled beta2 integrin-induced macrophage Rac2-Myosin IIA interaction regulates VEGF-A mRNA stability and arteriogenesis. J Exp Med. 2014;211:1957–68.
Kelley MJ, Jawien W, Ortel TL, Korczak JF. Mutation of MYH9, encoding non-muscle myosin heavy chain A, in May-Hegglin anomaly. Nat Genet. 2000;26:106–8.
Savoia A, De Rocco D, Panza E, Bozzi V, Scandellari R, Loffredo G, et al. Heavy chain myosin 9-related disease (MYH9 -RD): neutrophil inclusions of myosin-9 as a pathognomonic sign of the disorder. Thromb Haemost. 2010;103:826–32.
Schalkwijk J, Zweers MC, Steijlen PM, Dean WB, Taylor G, van Vlijmen IM, et al. A recessive form of the Ehlers-Danlos syndrome caused by tenascin-X deficiency. N Engl J Med. 2001;345:1167–75.
Grond-Ginsbach C, Debette S. The association of connective tissue disorders with cervical artery dissections. Curr Mol Med. 2009;9:210–4.
Debette S, Germain DP. Neurologic manifestations of inherited disorders of connective tissue. Handb Clin Neurol. 2014;119:565–76.
Lupski JR, Belmont JW, Boerwinkle E, Gibbs RA. Clan genomics and the complex architecture of human disease. Cell. 2011;147:32–43.
Hayashi T, Tanimoto K, Hirayama-Yamada K, Tsuda E, Ayusawa M, Nunoda S, et al. Genetic background of Japanese patients with pediatric hypertrophic and restrictive cardiomyopathy. J Hum Genet 2018;63:989–96.
Gbadegesin RA, Brophy PD, Adeyemo A, Hall G, Gupta IR, Hains D, et al. TNXB mutations can cause vesicoureteral reflux. J Am Soc Nephrol. 2013;24:1313–22.
Alcaraz LB, Exposito JY, Chuvin N, Pommier RM, Cluzel C, Martel S, et al. Tenascin-X promotes epithelial-to-mesenchymal transition by activating latent TGF-beta. J Cell Biol. 2014;205:409–28.
Acknowledgements
James R Lupski (Baylor College of Medicine) and Jennifer Posey (Baylor College of Medicine) assisted with organizing the study and revision of the manuscript. We thank the families who participated in this research. We also thank GeneSeeq Inc. for the technical support of whole-exome sequencing.
Author contributions
NW, XY, and PL conceived the project and designed the study. KW, QZ, XD, Jian Liu, XL, YZ, WC, and YN collected the cohorts. Sen Zhao, NW, PL, KW, QZ, MS, KA, JY, Jiachen Lin, and Bowen Liu analyzed the data. XS, YZ, YS, and ZY conducted the bioinformatic analysis. RD, Jiaqi Liu, BL, and BY helped with data interpretation and revised the manuscript. ZW, JS, and Shuyang Zhang helped in organizing the study and revising the manuscript. Sen Zhao, NW, KW, and PL wrote the manuscript.
Funding
Funding for this study was provided by grant no. 2016YFC1300800 from the National Key Research and Development Plan of China, nos. 81471167, 81671139, 81220108007, and no. 2014-1-1071 from the Special Research Project for Capital Health Development (all to Dr. Xinjian Yang), grant no. 81501852 from the National Natural Science Foundation of China; 2016 Milstein Medical Asian American Partnership Foundation Fellowship Award in Translational Medicine, no. 7172175 from the Beijing Natural Science Foundation, no. Z161100004916123 from the Beijing nova program, no. xxjc201717 from the Beijing nova program interdisciplinary collaborative project, no. 2016ZX310177 from the Central Level Public Interest Program for Scientific Research Institute, no. 3332016006 from the PUMC Youth Fund & the Fundamental Research Funds for the Central Universities, no. 2016-I2M-3-003 from the CAMS Initiative for Innovative Medicine, no. JQ201506 from the Distinguished Youth foundation of Peking Union Medical College Hospital (all to Dr. Nan Wu), and nos. 2016-I2M-2-006 and 2017-I2M-2-001 from the CAMS Initiative for Innovative Medicine (to Dr. Zhihong Wu).
Author information
Authors and Affiliations
Consortia
Corresponding authors
Ethics declarations
Ethical approval
This study was approved by the ethics committee of Beijing Tiantan Hospital. Informed consent was obtained from each patient or their parents.
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made.
The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this licence, visit https://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, K., Zhao, S., Zhang, Q. et al. Whole-exome sequencing reveals known and novel variants in a cohort of intracranial vertebral–basilar artery dissection (IVAD). J Hum Genet 63, 1119–1128 (2018). https://doi.org/10.1038/s10038-018-0496-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s10038-018-0496-x
This article is cited by
-
Delineation of dual molecular diagnosis in patients with skeletal deformity
Orphanet Journal of Rare Diseases (2022)
-
Phenotypic and genetic spectrum of isolated macrodactyly: somatic mosaicism of PIK3CA and AKT1 oncogenic variants
Orphanet Journal of Rare Diseases (2020)
-
A novel COMP mutation in a Chinese family with multiple epiphyseal dysplasia
BMC Medical Genetics (2020)
