
Abstract
Pediatric cardiomyopathy is a complex disease with clinical and genetic heterogeneity. Recently, the ALPK3 gene was described as a new hereditary cardiomyopathy gene underlying pediatric cardiomyopathies. Only eight patients carrying mutations in ALPK3 have been reported to date. Here, we report a 3-year-old male patient with both hypertrophic and dilated cardiomyopathy. The patient presented dysmorphic features and skeletal deformities of hands and feet, pectus excavatum, and cleft palate. The genetic investigation was performed by whole-exome sequencing in the patient and his parents. We identified a novel homozygous mutation in ALPK3 (c.1531_1532delAA; p.Lys511Argfs*12). Our work extends the phenotypic spectrum of the ALPK3-associated cardiomyopathy by reporting additional clinical features. This is the first study of a Tunisian patient with mutation in the ALPK3 gene. In conclusion, ALPK3 should be included in the list of genes to be considered in genetic studies for patients affected with pediatric syndromic cardiomyopathy.
Similar content being viewed by others


Introduction
Hypertrophic cardiomyopathy (HCM) and dilated cardiomyopathy (DCM) are the most common forms of primary cardiomyopathies and the major cause of heart failure and sudden cardiac death [1]. HCM is characterized by increased ventricular wall thickness associated with diastolic dysfunction of ventricular chambers. DCM is defined by dilation and systolic dysfunction of the left ventricle (LV) but right-ventricular involvement was also observed [1, 2]. These two entities occur in children with an annual incidence of 1.13 cases per 100.000 children younger than 18 years and 8.34 cases per 100.000 infants [3]. Moreover, higher incidence was found in boys and infants under 1 year of age for both HCM and DCM [3]. HCM and DCM are the most frequent defects among pediatric probands (51% HCM and 30% DCM) [4]. These findings were consistent with the results of the Pediatric Cardiomyopathy Registry (PCMR) [3, 5]. HCM and DCM have genetic and phenotypic heterogeneity with incomplete penetrance and variable expressivity. Different cardiac phenotypes are caused by mutations in the same gene, and numerous genes can be responsible of the same cardiomyopathy [6]. Furthermore, pediatric cardiomyopathies have both genetic and non-genetic etiologies. The genetic make-up of pediatric cardiomyopathies is more diversified than in the adult population because additional syndromic and metabolic genetic causes can be implicated [7]. Hence, the identification of the mutation spectrum and causal associated-genes are challenging. The advent of next generation sequencing technologies, such as whole-exome sequencing (WES) allowed the identification of the genetic basis of pediatric cardiomyopathies. Using this technique and the homozygosity mapping, the alpha-kinase 3 (ALPK3) gene was recently discovered as a novel pediatric cardiomyopathy-associated gene [8]. Indeed, homozygous mutations in the ALPK3 gene were identified in 5 patients with pediatric cardiomyopathy. This study hypothesized that ALPK3 might control initial induction of differentiation and maturation of cardiomyocytes by regulating the expression of cardiac transcription factors such as HEY2 and HAND [8]. The ALPK3 gene encodes a 1907 amino acids protein, which contains three functional domains: one α-type protein kinase domain and two immunoglobin-like domains. ALPK3 appears to be involved in cardiomyocytes differentiation and heart development [9]. A recent study using human induced pluripotent stem cell showed that absence of ALPK3 results in abnormal intracellular calcium [10]. Here, we report a novel homozygous frameshift mutation in the ALPK3 gene in a 3-year-old boy affected with mixed HCM/DCM phenotype and dysmorphic features.
Materials and methods
This study was conducted according to the principles of the Declaration of Helsinki and to the ethical standards of the first author’s institutional review board (Registration number: IRB00005445, FWA00010074).
The parents provided their written informed consent to participate in this study. After informed consent was obtained, peripheral blood samples were collected from the affected child and his parents.
Genomic DNA was extracted from the samples according to standard techniques. Whole-exome sequencing (WES) was performed for the trio (the affected child, the father and the mother), by the Genomics and Bioinformatics Platform (GBiM) from the UMR 1251/Marseille Medical Genetics facility, using the NimbleGen SeqCap EZ MedExome kit (total design size 47 Mb) according to the manufacturer’s protocol (Roche Sequencing Solutions, Madison, USA). The SeqCap EZ MedExome kit targets the entire human exome with enhanced coverage of exons from medically relevant genes in Mendelian diseases. Enriched fragment libraries were sequenced on the Illumina NextSeq 500 platform (Illumina, San Diego, CA, USA) using a 150 bp paired-end sequencing protocol. Raw data were mapped to the built of the human genome (hg19) by using BWA 0.7.5. Variant calling was subsequently performed using GATK and annotation was done with ANNOVAR. All subsequent steps were performed using our in-house software for variant annotation and segregation VarAFT (http://varaft.eu). WES data from the patient and his parents were analyzed simultaneously and segregated using VarAFT. All modes of inheritance have been tested during the analysis of the data.
In order to reduce the significant number of variants generated by WES sequencing and to identify relevant variants, a strategy of filtering was adopted. Variants were selected as following: [1] variants with Minor Allele Frequency (MAF) ≥ 1% in the Exome Aggregation Consortium database (ExAC) (http://exac.broadinstitute.org/) were removed [2]. The remaining variants were filtered based on their type and genomic localization, thus, synonymous, intronic, variants in intergenic, 3′ and 5′ UTR regions were discarded [3]. The obtained variants’ list (including rare, nonsense, missense, splice site, and frameshift INDELs), was then subsequently filtered according to the in silico pathogenicity prediction. Thus, variants predicted as polymorphism according to UMD-Predictor (http://umd-predictor.eu/), SIFT (http://sift.jcvi.org/), PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/), and Mutation Taster (http://www.mutationtaster.org/) were excluded.
Results
Clinical presentation
The patient, a 3-year-old Tunisian boy, is the only child of apparently healthy first cousins aged 21 (mother) and 28 (father) years. He was born at term by spontaneous vaginal delivery following an uncomplicated pregnancy. His birth weight was 3550 g. The patient was hospitalized at birth for 2 months and a week because of respiratory distress. At 7 days, he was diagnosed with mixed HCM/DCM cardiomyopathy, and began treatment by captopril (25 mg), furosemide (40 mg) and carvedilol (6.25 mg). Echocardiography performed at the age of 3 months revealed levocardia, situs solitus, LV dilation and concentric hypertrophy with altered systolic function (end-diastolic diameter (EDD) = 26 mm; end-systolic diameter (ESD) = 24 mm; inter-ventricular septum thickness at end-diastole (IVSd) = 9 mm; ejection fraction (EF) = 35%). The right-ventricular walls were enlarged. A patent foramen ovale with left to right shunt and mild tricuspid insufficiency (systolic pulmonary arterial pressure = 30 mmHg) were also reported. Neither aortic insufficiency nor valvular aortic stenosis were observed. There was no isthmic coarctation of the aorta. Cardiac examination at 6 months showed less dilation of the LV with improvement of the systolic function and concentric hypertrophy of both LV and RV (EDD = 27 mm; ESD = 20 mm; IVSd = 12 mm, EF = 50%). Minimal mitral valve insufficiency was noted. Echocardiographic indices at 8 months were as following: EDD = 26 mm; ESD = 19 mm; IVSd = 12 mm, EF = 52% and the LV posterior wall thickness = 12 mm. At 11 months, echocardiography revealed concentric hypertrophy of both ventricles without dilation (Fig. 1a.) (EDD = 27 mm; ESD = 15 mm; IVSd = 18 mm, EF = 74%). At 15 months, echocardiography showed a cardiomyopathy of predominantly hypertrophic LV without obstruction (Fig. 1b.). Abnormalities in left ventricular compliance were also observed and echocardiographic indices were as following: EDD = 25 mm; ESD = 16 mm; IVSd = 15 mm, EF = 65%, and the LV posterior wall thickness = 12 mm. Echocardiographic screening in the father revealed asymmetric septal hypertrophy (septal thickness = 16 mm) and minor congenital mitral anomaly without valvular dysfunction (Fig. 1c). The mother’s echocardiography was without abnormalities.

Echocardiographic images. a Echocardiography of the patient at the age of 11 months and b at 15 months. c Echocardiography of the father at the age of 30 years
First examination by geneticists at the age of 2 months and a half revealed a weight at 3800 g (<3rd centile), a height at 54 cm (<3rd centile) and a head circumference at 38.5 cm (3rd−15th centile), dysmorphic features including broad forehead, protruding eyes, divergent strabismus, blue sclera, low-set ears, long philtrum, thin lips, cleft palate, microretrognathia, and bilateral camptodactyly of hands and axial hypotonia.
On last examination, at the age of 2 years and 9 months, weight was on the 25th percentile; height and head circumference were on the 50th percentile. He had almost the same dysmorphic features characterized by flat occiput, triangular, and flat face, broad forehead, down-slanting palpebral fissures, protruding eyes, mild ptosis of the left eyelid, blue sclera, low set and backward-rotated small ears, broad nasal ridge and nasal tip, thick ala nasi, short columella, long philtrum, everted lips, cleft palate, microretrognathia, and full cheeks. He also had short neck, sloping shoulders, pectus excavatum, bilateral camptodactyly of hands, bilateral overlapping of the 2nd and 3rd toes, bilateral clinodactyly of the 4th and 5th toes, and knee stiffness. The psychomotor development of the patient was normal.
R-banded chromosome analysis on cultured peripheral blood lymphocytes from the proband was normal 46, XY. Fluorescent In Situ Hybridization (FISH) analysis with MD DiGeorge T-box1 (22q11)/22q13 (SHANK3) kreatech probe was performed but no microdeletion was identified. Creatine phosphokinase, lactate dehydrogenase, serum creatinine, ammonia, and lactates levels and chromatographic analysis of amino and organic acids were normal.
Whole-exome analysis
Taking into account the large number of genes mutated in inherited cardiac diseases we have performed a WES for the family (trio). WES data were analyzed and segregated using the Variant Annotation and Filtering Tool VarAFT. On average, 92 and 87% of all targeted exome was covered to a depth of 20× and 30×, respectively. All variants identified in our patient using the filtering strategy described above (Material and methods) are listed in Table 1 (homozygous mutations) and Supplementary Table 1 (heterozygous mutations). Based on these lists of variant we prioritized the homozygous frame-shift deletion in exon 5 of the ALPK3 gene (NM_020778): c.1531_1532delAA; p.Lys511Argfs*12 as a likely causative variant. Indeed, both parents were found heterozygous for this variant (Fig. 2). Moreover, the ALPK3 c.1531_1532delAA; p.Lys511Argfs*12 is not reported in the following databases: ExAC, dbSNP (https://www.ncbi.nlm.nih.gov/projects/SNP/), NHLBI Exome Sequencing Project (http://evs.gs.washington.edu/EVS/), gnomAD (http://gnomad.broadinstitute.org), 1000 Genomes (http://www.internationalgenome.org), and GME Variome (http://igm.ucsd.edu/gme/). In order to exclude the possibility that the ALPK3 p.Lys511Argfs*12 deletion could be a frequent variant specific of the Tunisian population, our in-house database, gathering WES data from 70 Tunisian individuals (140 chr) was queried and the variant was not present.

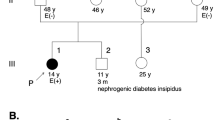
Pedigree of the family. Sanger electropherograms are shown below symbols. (+) indicates the wild-type allele. The arrow indicates the position of the mutation

A schematic representation of the ALPK3 gene. Blue boxes represent exons. Purple boxes represent untranslated regions (3′ and 5′ UTR). Mutations reported previously are indicated in blue color. Mutation identified in this study is indicated in red color. The three domains—Immunoglobulin-like (Ig-like, red boxes) and Alpha-kinase (green box) domains are also indicated
Discussion
We report a 3-year-old boy born to consanguineous parents of Tunisian origin and affected with mixed HCM/DCM, dysmorphic features, pectus excavatum, and skeletal deformities of both hands and feet.
Through an autosomal recessive model of inheritance analysis, we identified a novel frameshift deletion in ALPK3 not previously described in any frequency datasets. The ALPK3 c.1531_1532delAA variant is predicted to result in a premature stop codon (p.Lys511Argfs*12) leading to nonsense-mediated mRNA decay. To our knowledge, this novel variant in ALPK3 c.1531_1532delAA; is the sixth mutation identified in patient with pediatric cardiomyopathy, as only five mutations in ALPK3 have been reported to date: c.4736–1 G > A, c.3781 C > T, c.5294 G > A, c.3792 G > A, and c.2018delC (Fig. 3) [8, 10, 11].
The human ALPK3 gene (OMIM*617608) encodes the alpha-kinase 3 protein and is mainly expressed in heart and skeletal muscle. In 2001, Hosoda et al. identified the ALPK3 gene, also named MIDORI (murine Myocyte Induction Differentiation Originator) using the differential display technique in the mouse P19CL6 cell line system and demonstrated that its expression is enriched in the forming heart during early development.
Different studies suggested that ALPK3 is involved in the organization of myofibrils and the assembly of intercalated discs [8, 10, 12]. Indeed, a recent study using CRISPR/cas9 editing in a proband-derived iPSC line confirmed that the bi-allelic loss of function mutation in ALPK3 is pathogenic. Moreover, heterozygous correction of ALPK3 allowed the full restoration of the wild-type phenotype [10]. The loss of function of ALPK3 disrupts also membrane repolarization by extending the extracellular field potential duration. Calcium mishandling was also noted, which could underlie the cardiac hypertrophy and arrhythmia [10].
Mixed HCM/DCM phenotype was noted among children [6], in particular in patients mutated in the ALPK3 gene [8, 10, 11]. Previous studies have reported cases with progression of HCM to DCM [13,14,15]. Our patient displayed initially a mixed HCM/DCM phenotype that has evolved into a concentric hypertrophy of both left and right ventricles. Çağlayan et al. reported a 2½-year-old male patient carrying a homozygous frameshift mutation in ALPK3 (c.2018delC; p.Gln675Serfs*30) with a progression of DCM to HCM [11]. Moreover, three patients harboring mutations in ALPK3 displayed significant right-ventricular dysfunction [8]. On the cardiac level, the phenotype of our patient is consistent with that of patients carrying mutations in ALPK3 previously described [8, 10, 11].
In our study, both parents were found heterozygous for the ALPK3 c.1531_1532delAA; p.Lys511Argfs*12 mutation. Echocardiography of the mother did not show any cardiac dysfunction. However, the father presented hypertrophy of the inter-ventricular septum and minor congenital mitral anomaly. These observations are consistent with previous studies which found that heterozygous carriers of ALPK3 mutations displayed either no evidence of cardiomyopathy or later-onset cardiomyopathy or an atypical distribution of hypertrophy [8, 10].
Hence, the c.1531_1532delAA; p.Lys511Argfs*12 in ALPK3 at a homozygous state is the likely pathogenic mutation underlying the cardiac phenotype of our patient. Furthermore, we hypothesized that ALPK3 gene could be responsible of the dysmorphic and skeletal features of our patient. Low-set ears and high arched palate were the unique dysmorphic features described in one patient harboring ALPK3 mutation [11]. The clinical presentation of our patient is more diversified including cleft palate, pectus excavatum, bilateral camptodactyly, bilateral clinodactyly, and knee stiffness. Tissues expression of all identified variants (Table 1 and Supplementary Table 1) using GTEx database as well as related OMIM diseases did not reveal an association with skeletal deformities, pectus excavatum and cleft palate, except the ALPK3 variant. Indeed, previous studies in the mouse revealed that ALPK3 is expressed in the fetal and adult heart, as well as branchial arch and skeletal muscles [9, 12]. Similarly, a high level of ALPK3 transcript was detected in cardiac and skeletal muscles in humans [10].
In conclusion, our study reinforces the implication of ALPK3 in pediatric cardiomyopathy and sheds light on the syndromic nature of the disease. Therefore, we have extended the mutational and phenotypic spectrum of ALPK3-related disease by reporting a novel homozygous mutation associated with additional clinical features.
References
Kimura A. Molecular genetics and pathogenesis of cardiomyopathy. J Hum Genet. 2016;61:41.
Mestroni L, Rocco C, Gregori D, Sinagra G, Di Lenarda A, Miocic S, et al. Familial dilated cardiomyopathy: evidence for genetic and phenotypic heterogeneity. Heart Muscle Disease Study Group. J Am Coll Cardiol. 1999;34:181–90.
Wilkinson JD, Sleeper LA, Alvarez JA, Bublik N, Lipshultz SE. The Pediatric Cardiomyopathy Registry: 1995–2007. Prog Pediatr Cardiol. 2008;25:31–6.
Kindel SJ, Miller EM, Gupta R, Cripe LH, Hinton RB, Spicer RL, et al. Pediatric cardiomyopathy: importance of genetic and metabolic evaluation. J Card Fail. 2012;18:396–403.
Wilkinson JD, Westphal JA, Bansal N, Czachor JD, Razoky H, Lipshultz SE. Lessons learned from the Pediatric Cardiomyopathy Registry (PCMR) Study Group. Cardiol Young. 2015;25(Suppl 2):140–53.
Lee TM, Hsu DT, Kantor P, Towbin JA, Ware SM, Colan SD, et al. Pediatric cardiomyopathies. Circ Res. 2017;121:855–73.
Mestroni L, Sweet ME, Taylor MRG. Pediatric cardiomyopathy. J Am Coll Cardiol. 2016;67:526–8.
Almomani R, Verhagen JMA, Herkert JC, Brosens E, van Spaendonck-Zwarts KY, Asimaki A, et al. Biallelic truncating mutations in ALPK3 cause severe pediatric cardiomyopathy. J Am Coll Cardiol. 2016;67:515–25.
Hosoda T, Monzen K, Hiroi Y, Oka T, Takimoto E, Yazaki Y, et al. A novel myocyte-specific gene Midori promotes the differentiation of P19CL6 cells into cardiomyocytes. J Biol Chem. 2001;276:35978–89.
Phelan DG, Anderson DJ, Howden SE, Wong RCB, Hickey PF, Pope K, et al. ALPK3-deficient cardiomyocytes generated from patient-derived induced pluripotent stem cells and mutant human embryonic stem cells display abnormal calcium handling and establish that ALPK3 deficiency underlies familial cardiomyopathy. Eur Heart J. 2016;37:2586–90.
Çağlayan AO, Sezer RG, Kaymakçalan H, Ulgen E, Yavuz T, Baranoski JF, et al. ALPK3 gene mutation in a patient with congenital cardiomyopathy and dysmorphic features. Mol Case Stud. 2017;3:a001859.
Van Sligtenhorst I, Ding Z-M, Shi Z-Z, Read RW, Hansen G, Vogel P. Cardiomyopathy in α-kinase 3 (ALPK3)–deficient mice. Vet Pathol. 2012;49:131–41.
Spirito P, Maron BJ. Absence of progression of left ventricular hypertrophy in adult patients with hypertrophic cardiomyopathy. J Am Coll Cardiol. 1987;9:1013–7.
Hecht GM, Klues HG, Roberts WC, Maron BJ. Coexistence of sudden cardiac death and end-stage heart failure in familial hypertrophic cardiomyopathy. J Am Coll Cardiol. 1993;22:489–97.
Freeman K, Colon-Rivera C, Olsson MC, Moore RL, Weinberger HD, Grupp IL, et al. Progression from hypertrophic to dilated cardiomyopathy in mice that express a mutant myosin transgene. Am J Physiol Heart Circ Physiol. 2001;280:H151–9.
Acknowledgements
We thank the patient's family for their collaboration. This work was supported by the Tunisian Ministry of Public Health, the Ministry of Higher Education and Scientific Research (LR16IPT05). The project leading to this publication has received funding from Excellence Initiative of Aix-Marseille University - A*MIDEX, a French “Investissements d’Avenir” programme (RARE-MED project).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Jaouadi, H., Kraoua, L., Chaker, L. et al. Novel ALPK3 mutation in a Tunisian patient with pediatric cardiomyopathy and facio-thoraco-skeletal features. J Hum Genet 63, 1077–1082 (2018). https://doi.org/10.1038/s10038-018-0492-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s10038-018-0492-1
This article is cited by
-
Exploring the complex spectrum of dominance and recessiveness in genetic cardiomyopathies
Nature Cardiovascular Research (2023)
-
Compound Heterozygosity for Late-Onset Cardiomyopathy-Causative ALPK3 Coding Variant and Novel Intronic Variant Cause Infantile Hypertrophic Cardiomyopathy
Journal of Cardiovascular Translational Research (2023)
-
Minor hypertrophic cardiomyopathy genes, major insights into the genetics of cardiomyopathies
Nature Reviews Cardiology (2022)
