
Abstract
Argininosuccinic aciduria (ASA), which is considered to be the second most common urea cycle disorder (UCD), is caused by an argininosuccinate lyase deficiency and is biochemically characterized by elevation of argininosuccinic acid and arginine deficiency. In addition to hyperammonemia, other characteristic features of ASA include hepatic fibrosis, hypertension, neurocognitive deficiencies, and trichorrhexis nodosa. Herein, we retrospectively reviewed the clinical findings, biochemical profiles, and genotypic characteristics of five Korean patients with ASA, who showed typical phenotypes and biochemical findings of the disease. Molecular analysis of these patients revealed six novel ASL mutations. Next, we investigated the prevalence of all types of UCDs in Korea. Of note, over a two decade periods, ASA was only detected in 6.3% of patients with a UCD, which made it the fourth most common UCD in Korea. In comparison with Caucasians, in whom ASA is the second most common UCD, ASA is comparatively rare in East Asian populations, including Japanese and Koreans. These findings suggest the possibility of geographic variation in UCDs among ethnic groups.
Similar content being viewed by others

Introduction
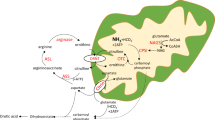
Argininosuccinic aciduria (ASA, OMIM #207900) is a urea cycle disorder (UCD) caused by a deficiency in agininosuccinate lyase (ASL). ASA is inherited in an autosomal recessive manner, and its incidence rate is estimated at 1:70,000 live births. ASA is the second most common UCD worldwide [1]. In Korea, more than 80% of newborns are screened for inherited metabolic disorders as part of the newborn screening program by tandem mass spectrometry [2]. An elevation of citrulline at newborn screening suggests that the baby might be affected by citrullinemia type 1 (CTLN1), ASA, or citrullinemia type 2 (CTLN2; citrin deficiency), further biochemical, molecular, and/or enzymatic testing is required for confirmatory diagnosis [3, 4]. The clinical manifestation of ASA is classified into two groups: a severe neonatal-onset form, presenting hyperammonemia within the first few days after birth and a late-onset form, with or without obvious episodic hyperammonemia. ASA is characterized by long-term complications, including chronic liver dysfunction, neurocognitive dysfunction, trichorrhexis nodosa, electrolyte imbalance, and hypertension [3]. Previously, our group reported various disease conditions as UCDs in Korea [5,6,7,8]. In this study, we reviewed the phenotypic, genotypic, and biochemical profiles and the treatment outcomes of ASA patients in Korea. We also determined the prevalence of ASA among inherited UCDs in Korea and compared these data to that in other populations.
Materials and methods
Patients
Five patients were included in our study. The diagnosis of ASA was based on biochemical findings and molecular analyses of the ASL gene (NM 000048). We retrospectively reviewed the clinical findings, including age at diagnosis, presenting symptoms, and developmental outcomes. In addition, biochemical findings, including plasma ammonia, plasma amino acid, and plasma and/or urine argininosuccinic acid levels were assessed. Written informed consent was obtained from all study subjects or their parents. The study protocol was approved by the Institutional Review Board of Asan Medical Center, Seoul, Korea.
Amino acid analysis
Serum urine and amino acid levels were analyzed by liquid chromatography-tandem mass spectrometry. An aliquot of 40 μL of the sample was added to 10 μL of 10% sulfosalicylic acid to precipitate proteins. After mixing and centrifugation (10,000×g for 2 min), the supernatant was mixed with 40 μL of borate buffer. Next an aliquot of 10 μL of the obtained solution was derivatized with the reagents supplied in the aTRAQ Kit for Amino Acid Analysis (Sciex, Framingham, MA, USA). The samples labeled with aTRAQ reagent Δ8 were mixed with the internal standards pre-labeled with aTRAQ reagent Δ0. The determination of free amino acid levels was conducted using an HPLC instrument 1200 Infinity (Agilent Technologies, Santa Clara, CA, USA) combined with a 3200 Q-TRAP mass spectrometer (Sciex) with an electrospray ionization source.
Molecular analysis of the ASL gene
Genomic DNA was extracted from peripheral blood leukocytes with the Gentra Puregene blood kit (Qiagen, Hilden, Germany). Direct sequencing of the ASL gene was performed using the extracted genomic DNA. All 17 coding exons and the exon–intron boundaries of the genes of interest were individually amplified by polymerase chain reaction (PCR) using primers designed from the flanking regions. Amplified PCR products were directly sequenced by using the BigDye Terminator v.3.1 Cycle Sequencing Kit and an ABI3130x1 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA). A real-time genomic PCR assay was performed to identify the copy number of each ASL exon(s), using 16 sets of primer pairs. Each primer pair was designed to be located in each exon. We used genomic DNA from two normal females as controls, and real-time PCR analyses with each primer pair were repeated three times. ASL consists of 17 exons and coding region includes exons 2–17. Therefore, we used the 16 sets of primer pairs for PCR (Table 1).
Spectrum of UCDs in a South Korean population
Patients who were diagnosed with each UCD such as ASA, ornithine transcarbamylase (OTC) deficiency, CTLN1, carbamoyl phosphate synthetase1 (CPS1) deficiency, N-acetylglutamate synthase (NAGS) deficiency, hyperammonemia-hyperornithinemia-homocitrullinuria (HHH) syndrome, and argininemia, at Asan Medical Center, Seoul, Korea from January 1999 to December 2017, were included in the current study. The number of patients with each UCD was calculated.
Results
Clinical features of ASA patients at presentation
Among the five included ASA patients (three males and two females, numbered 1–5), patients 1 and 2 had early onset ASA, which presented as hyperammonemic encephalopathy, whereas patients 3–5 had the late-onset form, which was identified by newborn screening (Table 2). Patient 1 manifested tachypnea and lethargy with hyperammonemia (344 µmol/L) and patient 2 developed comatose mentality and poor oral intake due to extremely high blood ammonia (988 µmol/L) a few days after birth. Patients 3–5 were given medical attention due to high blood citrulline levels at newborn screening, which was performed 2–3 days after birth. The ammonia levels of patients 3–5 were normal or only mildly elevated (45–104 µmol/L). Emergent management was required for patients 1 and 2, and they were administered intravenous sodium benzoate and oral phenylbutyrate and arginine to ameliorate hyperammonemia. Patient 2 required continuous venovenous hemodiafiltration for 24 h to rapidly correct hyperammonemia. At diagnosis, patients 1 and 2 showed elevation of glutamine level, and normal range of arginine level. Also patient 1 showed elevation of glutamate level. Patients 3–5 showed normal range of glutamine, glutamate, and arginine (Table 2). Patients 1–4 showed elevated argininosuccinic acid levels in serum and, or urine (Table 2). Patient 5 had no elevation of argininosuccinic acid at initial serum amino acid analysis, but its elevation was detected for the first time at 16 months of age, although the exact value was not available. An electrolyte imbalance was noted in patient 2, who had hypokalemia with increased renal excretion (transtubular potassium gradient, 17.19).
Mutational analysis
A total of eight different mutations were identified in the 10 tested alleles (Table 3). Two mutations (p.Arg182Gln, p.Arg186Gln) were previously reported [9, 10], while the other six were novel mutations (p.Lys287Asn, p.Leu419Argfs*6, p.Trp137*, p.Tyr375Cys, p.Pro156Leu, and an exon 15 deletion). One mutation, p.Pro156Leu was found in three alleles. According to the Genome Aggregation Database (gnomAD), the minor allele frequency of the c.467C>T (p.Pro156Leu) variant is 0.000008136. The p.Pro156Leu mutation was predicted to be pathogenic by several in silico prediction programs, including PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/), Sorting Intolerant From Tolerant (http://sift.jcvi.org/), and MutationTaster (http://www.mutationtaster.org/). Two other novel mutations, p.Lys287Asn and p.Tyr375Cys, were not observed in the control population by gnomAD (http://gnomad.broadinstitute.org/), ExAc (http://exac.broadinstitute.org/), KRGDB (http://152.99.75.168/KRGDB/menuPages/intro.jsp/), and were also predicted to be pathogenic by in silico prediction programs. p.Leu419Argfs*6 and p.Trp137* were novel frameshift and nonsense mutations. Exon 15 deletion was detected by genomic real-time PCR analysis.
Clinical courses of the ASA patients
All five patients have continued maintenance therapy. They have been managed with a protein-restricted diet (1.0–1.5 g/kg/day) and arginine supplementation (100–150 mg/kg/day). Plasma levels were monitored on a regular basis. In addition, phenylbutyrate (100–200 mg/kg/day) and sodium benzoate (100–150 mg/kg/day) were administered to prevent hyperammonemic episodes. Under close clinical observation, patient 3 was able to discontinue the nitrogen scavenging therapies without a hyperammonemic episode 3 years of age. The Korean Developmental Test for infants and toddlers was conducted for all patients [11]. In this test, a score of >80 in each of five domains including gross motor, fine motor, personal-social, language, and cognitive-adaptive skills indicates normal development. Patients 1 and 2 with neonatal-onset scored in the 50–60 s in all domains until the latest evaluation. Among patients with late-onset ASA (patients 3–5), only patient 4 had a score in the 60 s in fine motor skill and language development until 2 years of age. All five patients presented a normal range of AST/ALT level. Additionally, their abdominal ultrasonography findings were normal except in patients 1 and 2. Patients 1 and 2 developed hepatomegaly based on physical examination and ultrasonography. Patients 1, 3, and 4 had brittle, sparse hair. All patients had normal blood pressure during the 2–7 years of observation.
Spectrum of UCDs in a Korean population
Eighty patients were diagnosed with a UCD in the Medical Genetics Center of Asan Medical Center, Seoul, Korea, by biochemical and molecular analyses, during the period from January 1999 to December 2017. The entire spectrum of UCDs diagnosed at our institution was evaluated to determine the prevalence in our UCD cohort, and only five patients were diagnosed with ASA. The most common disorder was OTC deficiency (36 patients, 45.0%), and the second and third most common UCDs were CTLN1 (25 patients, 31.3%) and CPS1 deficiency (11 patients, 13.8%), respectively. In contrast, only 5 patients (6.3%) were diagnosed with ASA, while 1.3% (each) were diagnosed with argininemia, HHH, and NAGS deficiency (Fig. 1).

Discussion
The current study is the first to describe the clinical and molecular characteristics of ASA patients in a Korean population. The five patients described here showed the typical phenotypes and biochemical characteristics of ASA. Two patients were affected with neonatal-onset severe ASA, while the remaining three patients had less severe late-onset ASA. The distinct clinical features of ASA have been described as a constellation of phenotypes, including trichorrhexis nodosa, electrolyte imbalance (hypokalemia), hypertension, hepatic disease, and neurocognitive deficiency. Neurodevelopmental delay is also found in other UCDs, but is more common in patients with ASA [3]. Hair is composed of 10.5% arginine by weight, thus the insufficient arginine of the patients with ASA results in weak hair shaft [12]. In this study, three patients presented trichorrhexis nodosa, which was improved with arginine supplementation. The mechanism of hypokalemia in ASA is unclear; however, increased renal loss may be involved [3]. In our study, one patient has presented hypokalemia with increased renal potassium excretion. The severity of hepatic dysfunction in ASA is variable, ranging from hepatomegaly to liver cirrhosis. The hepatic disease is considered to be progressive [13,14,15,16], and hepatic fibrosis is often associated in children with neonatal-onset ASA [17, 18]. Although, there were only two patients with hepatomegaly in our study, based on short-term follow-up period for each patient, life-long monitoring is needed. Neurocognitive deficits are often observed more frequently in patients with neonatal-onset ASA than in patients with late-onset ASA, as was also observed for our patients. Neurological developmental delay is associated with the ammonia level in a hyperammonemic episode, the glutamine level, and the duration of hyperammonemic exposure. Thus, early identification and intervention are very important to improve neurological outcomes. Although, late-onset ASA patients may never have a hyperammonemic events, like patient 4 [19], neurocognitive deficits can still develop. Therefore, long-term observation for neurocognitive deficits is required in all patients with ASA, and early intervention is required to minimize these deficits. All patients with ASA in Korea receive arginine supplementation, are put on a protein-restricted diet and are treated with ammonia scavengers. Although, sodium nitrite is not yet available in Korea, sodium nitrite therapy is expected to become a standard treatment for ASA in the near future to help prevent long-term complications [15, 20, 21]. On a molecular level, it is notable that six out of the seven ASL mutations identified in the five patients were novel and that p.Pro156Leu was present in three alleles of two unrelated patients. Two mutations in our ASA patients, p.Arg182Gln and p.Arg186Gln, were previously reported in Dutch and Italian patients, respectively [9, 10]. In Caucasians, p.Arg385Cys, p.Gln116Ter, p.Arg12Gln, p.Ile100Thr, p.Arg186Trp, p.Glu189Gly, p.Gln286Arg, and p.Val178Met are common mutations [10, 22, 23], but are not found in Korea. Unfortunately, we could not find an appropriate article describing the ASL mutation spectrum in Japan or other Asian countries. Additional studies are needed to characterize the molecular genetic features of ASA among different ethnicities. As a major tertiary medical center in Korea, our center has been taking care of most of UCD patients in Korea [5,6,7,8], therefore, the proportions of each UCD diagnosed in our center likely represent the spectrum of UCDs in Korea. When we compared the relative prevalence of each UCD in Korea to those in Japan, the USA, and European countries, a difference was observed among these populations (Fig. 1). OTC deficiency was the most common phenotype worldwide. However, ASA is the second most common inherited UCD in the USA and European countries [14, 24, 25], whereas ASA was as rare as 5.1–5.6% in Japan and 6.3% in Korea, making it the fourth most common UCD in these East Asian countries [26, 27]. We also investigated the spectrum of UCDs in other East Asian countries, including China and Taiwan. Although, there were no epidemiological surveys of UCDs, ASA is expected to be extremely rare in these countries as well [28, 29]. In contrast, ASA was the most common UCD in Malaysia [30, 31]. These findings suggest that there might exist the ethnic differences in the prevalence rates of each UCD among various populations. In conclusion, ASA is expected to be rare in the perspective of the UCD spectrum in East Asian countries, suggesting geographic variation in UCDs among ethnic groups. Further studies are required to identify more cases in diverse populations to understand the clinical and molecular spectrum of ASAs and share the clinical experiences to develop new treatment strategies.
References
Erez A. Argininosuccinic aciduria: from a monogenic to a complex disorder. Genet Med. 2013;15:251–7.
Cho SE, Park EJ, Seo DH, Lee IB, Lee HJ, Cho D-Y, et al. Neonatal screening tests for inherited metabolic disorders using tandem mass spectrometry: experience of a clinical laboratory in Korea. Lab Med Online. 2015;5:196–203.
Nagamani SC, Erez A, Lee B. Argininosuccinate lyase deficiency. Genet Med. 2012;14:501–7.
Wilcken B, Wiley V, Hammond J, Carpenter K. Screening newborns for inborn errors of metabolism by tandem mass spectrometry. N Engl J Med. 2003;348:2304–12.
Choi JH, Lee BH, Kim JH, Kim GH, Kim YM, Cho J, et al. Clinical outcomes and the mutation spectrum of the OTC gene in patients with ornithine transcarbamylase deficiency. J Hum Genet. 2015;60:501–7.
Kim JH, Kim YM, Lee BH, Cho JH, Kim GH, Choi JH, et al. Short-term efficacy of N-carbamylglutamate in a patient with N-acetylglutamate synthase deficiency. J Hum Genet. 2015;60:395–7.
Lee BH, Kim YM, Heo SH, Kim GH, Choi IH, Lee BS, et al. High prevalence of neonatal presentation in Korean patients with citrullinemia type 1, and their shared mutations. Mol Genet Metab. 2013;108:18–24.
Lee BH, Jin HY, Kim GH, Choi JH, Yoo HW. Argininemia presenting with progressive spastic diplegia. Pediatr Neurol. 2011;44:218–20.
Trevisson E, Salviati L, Baldoin MC, Toldo I, Casarin A, Sacconi S, et al. Argininosuccinate lyase deficiency: mutational spectrum in Italian patients and identification of a novel ASL pseudogene. Hum Mutat. 2007;28:694–702.
Linnebank M, Tschiedel E, Haberle J, Linnebank A, Willenbring H, Kleijer WJ, et al. Argininosuccinate lyase (ASL) deficiency: mutation analysis in 27 patients and a completed structure of the human ASL gene. Hum Genet. 2002;111:350–9.
Kim J-H, Yum M-S, Jeong S-J, Ko T-S. Assessment of children with developmental delay: Korean infant and child development test (KICDT) and Korean Bayley scale of infant development-II (K-BSID-II). Korean J Pediatr. 2009;52:772–7.
Fichtel JC, Richards JA, Davis LS. Trichorrhexis nodosa secondary to argininosuccinicaciduria. Pediatr Dermatol. 2007;24:25–7.
Widhalm K, Koch S, Scheibenreiter S, Knoll E, Colombo JP, Bachmann C, et al. Long-term follow-up of 12 patients with the late-onset variant of argininosuccinic acid lyase deficiency: no impairment of intellectual and psychomotor development during therapy. Pediatrics. 1992;89:1182–4.
Mercimek-Mahmutoglu S, Moeslinger D, Haberle J, Engel K, Herle M, Strobl MW, et al. Long-term outcome of patients with argininosuccinate lyase deficiency diagnosed by newborn screening in Austria. Mol Genet Metab. 2010;100:24–28.
Mori T, Nagai K, Mori M, Nagao M, Imamura M, Iijima M, et al. Progressive liver fibrosis in late-onset argininosuccinate lyase deficiency. Pediatr Dev Pathol. 2002;5:597–601.
Nagamani SC, Campeau PM, Shchelochkov OA, Premkumar MH, Guse K, Brunetti-Pierri N, et al. Nitric-oxide supplementation for treatment of long-term complications in argininosuccinic aciduria. Am J Hum Genet. 2012;90:836–46.
Ficicioglu C, Mandell R, Shih VE. Argininosuccinate lyase deficiency: longterm outcome of 13 patients detected by newborn screening. Mol Genet Metab. 2009;98:273–7.
Baruteau J, Jameson E, Morris AA, Chakrapani A, Santra S, Vijay S, et al. Expanding the phenotype in argininosuccinic aciduria: need for new therapies. J Inherit Metab Dis. 2017;40:357–68.
Brunetti-Pierri N, Erez A, Shchelochkov O, Craigen W, Lee B. Systemic hypertension in two patients with ASL deficiency: a result of nitric oxide deficiency? Mol Genet Metab. 2009;98:195–7.
Nagamani SC, Shchelochkov OA, Mullins MA, Carter S, Lanpher BC, Sun Q, et al. A randomized controlled trial to evaluate the effects of high-dose versus low-dose of arginine therapy on hepatic function tests in argininosuccinic aciduria. Mol Genet Metab. 2012;107:315–21.
Nagamani SC, Lee B, Erez A. Optimizing therapy for argininosuccinic aciduria. Mol Genet Metab. 2012;107:10–4.
Keskinen P, Siitonen A, Salo M. Hereditary urea cycle diseases in Finland. Acta Paediatr. 2008;97:1412–9.
Balmer C, Pandey AV, Rufenacht V, Nuoffer JM, Fang P, Wong LJ, et al. Mutations and polymorphisms in the human argininosuccinate lyase (ASL) gene. Hum Mutat. 2014;35:27–35.
Tuchman M, Lee B, Lichter-Konecki U, Summar ML, Yudkoff M, Cederbaum SD, et al. Cross-sectional multicenter study of patients with urea cycle disorders in the United States. Mol Genet Metab. 2008;94:397–402.
Summar ML, Koelker S, Freedenberg D, Le Mons C, Haberle J, Lee HS, et al. The incidence of urea cycle disorders. Mol Genet Metab. 2013;110:179–80.
Uchino T, Endo F, Matsuda I. Neurodevelopmental outcome of long-term therapy of urea cycle disorders in Japan. J Inherit Metab Dis. 1998;21:151–9. Suppl 1
Nakamura K, Kido J, Mitsubuchi H, Endo F. Diagnosis and treatment of urea cycle disorder in Japan. Pediatr Int. 2014;56:506–9.
Han LS, Ye J, Qiu WJ, Gao XL, Wang Y, Gu XF. Selective screening for inborn errors of metabolism on clinical patients using tandem mass spectrometry in China: a four-year report. J Inherit Metab Dis. 2007;30:507–14.
Niu DM, Chien YH, Chiang CC, Ho HC, Hwu WL, Kao SM, et al. Nationwide survey of extended newborn screening by tandem mass spectrometry in Taiwan. J Inherit Metab Dis. 2010;33:S295–S305.
Chen BC, Ngu LH, Zabedah MY. Argininosuccinic aciduria: clinical and biochemical phenotype findings in Malaysian children. Malays J Pathol. 2010;32:87–95.
Thong MK, Yunus ZM. Spectrum of inherited metabolic disorders in Malaysia. Ann Acad Med Singapore. 2008;37:66–65.
Acknowledgements
This study was supported in part by a grant from the National Research Foundation of Korea, funded by the Ministry of Education, Science, and Technology (NRF-2016M3A9B4915706).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Kim, D., Ko, J.M., Kim, Ym. et al. Low prevalence of argininosuccinate lyase deficiency among inherited urea cycle disorders in Korea. J Hum Genet 63, 911–917 (2018). https://doi.org/10.1038/s10038-018-0467-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s10038-018-0467-2
