
Abstract
We report a Thai father (patient 1) and his daughter (patient 2) affected with osteogenesis imperfecta type IV and dentinogenesis imperfecta. Both were heterozygous for the c.1451G>A (p.Gly484Glu) mutation in COL1A2. The father, a Thai boxer, had very mild osteogenesis imperfecta with no history of low-trauma bone fractures. Scanning electron micrography of the primary teeth with DI of the patient 2, and the primary teeth with DI of another OI patient with OI showed newly recognized dental manifestations of teeth with DI. Normal dentin and cementum might have small areas of ectopic mineralizations. Teeth affected with DI have well-organized ectopic mineralizations in dentin and cementum. The “French-fries-appearance” of the crystals at the cemento-dentinal junction and abnormal cementum have never been reported to be associated with dentinogenesis imperfecta, either isolated or osteogenesis imperfecta-associated. Our study shows for the first time that abnormal collagen fibers can lead to ectopic mineralization in dentin and cementum and abnormal cementum can be a part of osteogenesis imperfecta.
Similar content being viewed by others
Introduction
Osteogenesis imperfecta (OI) is a phenotypically and molecularly heterogeneous group of inherited connective tissue disorders characterized by skeletal abnormalities with high susceptibility to bone fragility and deformity. Severity of the phenotypes ranges from perinatal death to barely unnoticeable clinical features. Most cases of OI are caused by mutations in two prominent collagen genes, COL1A1 (MIM #120150) and COL1A2 (MIM #120160), responsible for producing pro-α1(I) and pro-α1(II) chains, respectively. The majority of the previously reported mutations were glycine residue substitutions in the triple helix domain of the encoded protein [1, 2]. Phenotypes of OI are known to vary even within the same families. Herein, we report a father and his daughter, both affected with mild OI and dentinogenesis imperfecta (DI). Scanning electron micrographs (SEM) of teeth with DI showed newly recognized dental manifestations, bush-like ectopic mineralizations in dentin and cementum. This report shows varying clinical features of patients with OI. Furthermore, a patient with OI may have ectopic mineralization in dentin and cementum, and no bone fragility.
Materials and methods
Ethic statement
The study was conducted in accordance with the Declaration of Helsinki and national guidelines. Informed consent was obtained from the patient and her parent in accordance with the regulations of the Human Experimentation Committee of the Faculty of Dentistry, Chiang Mai University.
Patient 1
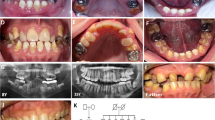
A 36-year-old Thai male and his four-year old daughter came to the Pediatric Dental Clinic, Faculty of Dentistry, Chiang Mai University for dental evaluation. His height, weight, and head circumference were 151 cm (<3rd centile), 58 kg (50th centile), and 57 cm (50th centile), respectively. Disproportionate short stature was evident with the lower part of the body disproportionately shorter than the upper part (upper to lower segment ratio 1.1:1, and arm span 144 cm) (Fig. 1a). Radiographic examination showed generalized mild osteopenia (Fig. 2a–g). Slight concave deformity of the endplates was observed (Fig. 2c). He was a Thai boxer between the ages of 13 and 15 years. At the age of 17, he experienced a fracture of the left scapular body and dislocation of the left shoulder due to a car accident (Fig. 3c). Dual-energy x-ray absorptiometry of the L1–L4 regions at age 36 years showed low bone mineral density (BMD) (0.697 g/cm2; T-score of –3.6; age-matched Z-score of –3.2). Oral examination showed DI of the permanent teeth. A number of teeth were lost as a result of dental infection (Fig. 1c). The patient could not give a definite dental history. A panoramic radiograph showed absence of a number of permanent teeth, pulp obliteration, bulbous-shaped permanent molars, and dense mandibular bone. The roots of all his teeth appeared shorter than normal (Fig. 3d).

Patient 1. a, c A Thai boxer affected with OI and DI. His legs are disproportionately short. A number of teeth were lost as a result of dental infections. Patient 2. b, d The four-year-old daughter of Patient 1 also affected with OI and DI. Maxillary incisors have severe dental caries and dental infections. e Normal incisor and incisors affected with DI. Note incisors with DI appear translucent

Radiographs of Patient 1. a–g Generalized mild osteopenia. c Mild ostepenia of the vertebrae with slight concave deformity of the endplates. In general, his skeletal malformations are not remarkable

Radiographs of Patient 1. a A-P hand radiograph. Note ivory epiphyses of the proximal phalanges of hands. b A-P radiograph of feet. Ivory epiphyses of the proximal phalanges are noted. c Dislocation of the left shoulder. d Panoramic radiograph of Patient 1. Note dense mandibular bone. A number of teeth are lost. Short tooth roots and dental pulp obliteration are noted. e Panoramic radiograph of Patient 2. Primary teeth and developing permanent teeth are observed. Bulbous molars. Large pulp chambers and root canal spaces. Dentin of the developing mandibular first molars is not evident
Patient 2
A 4-year-old girl, the daughter of Patient 1 also had disproportionate short stature with the lower part of the body disproportionately shorter than the upper part (Fig. 1b). At birth, her upper to lower segment ratio, arm span, and occipitofrontal circumference were 2.2:1, 41 cm with length 45 cm (<3rd centile), and 34 cm (50th centile), respectively. Her current height, weight, and OFC were 89 cm (<3rd centile), 12 kg (3rd centile), and 50 cm (75th centile), respectively. The left leg appeared curved (Fig. 1b). Radiographic examination at age 2 days showed mild osteopenia of the cranium with islands of wormian bones (Fig. 4c). Long bones were slender, curved, and slightly osteopenic. Metaphyses of the femora, tibiae, and humeri appeared enlarged. Shortened proximal radii were noted (Fig. 4d–g). She sustained a left femoral fracture at age 1 ½ years, due to a fall in a bathroom. At age 3 years and 9 months the humeri, femora, and tibiae were curved with enlarged metaphyses. Fracture of the left tibia as a result of a fall was noted at age 4 years. Distal epiphyses of the femora and proximal epiphyses of the tibiae were thickened. The fibulae appeared very thin. Collapsed T11–12 vertebral bodies were identified. Generalized slight osteopenia with longitudinal striation was observed. At the time of this study, her osteopenia was less severe than that after birth (Fig. 5a–d). Dual-energy x-ray absorptiometry of the L1–L4 regions at age 4 years showed low BMD (BMD ho0.332 g/cm2). Unfortunately, there are no standard BMD measurements for young ages or comparison. At age 3 years, pamidronate treatment with dosage of 9 mg/kg/year was provided every 4 months. Nine months after pamidronate treatment was begun, she sustained a left femoral fracture again.

Radiographs of Patient 2. At age 2 days. Note generalized osteopenia. a A-P and b Lateral full length view of the spine. Note generalized osteopenia of slender bones. Femora are curved with enlarged metaphyses. c Lateral skull radiograph at age 2 days. Wormian bones are noted (arrow). d, e Curved and enlarged metaphyses of femora and tibiae. Short proximal radii. Carpal bones are not visible. f, g At age 8 months. Compared to the bones at age 2 days, bones are less osteopenic. Proximal radius is short. Curved and enlarged metaphyses of humeri, femora, and tibiae. Capitate and hamate bones are visible

Radiographs taken at ages 3 years and 9 months. a–d Improvement of osteopenia is noted. Longitudinal striation of long bone as a result of osteopenia is observed. b The shortening of proximal radii is improved. Enlargement of proximal humeri is less obvious. d Curved femora and fracture of left tibia are noted (arrow)
Oral examination showed DI of the primary dentition. Severe dental caries and pulp exposures were observed in the maxillary deciduous incisors (Fig. 1d). Periapical radiograph of the maxillary deciduous incisors showed retained roots with radiolucent areas at their periapical regions, indicating dental infection. The radiopacity of the dentin was less than normal. The extracted maxillary incisors appeared thin and translucent (Fig. 1e). A panoramic radiograph recorded at age 4 years showed bulbous molars with large pulp chambers and root canal spaces (Fig. 3e).
Scanning electron micrography
Teeth that were studied under SEM included maxillary primary incisors affected with DI of Patient 2, maxillary primary incisors affected with DI of a patient with OI (Supplemental Figure S3), and normal teeth. All teeth were split using a dental chisel and hammer, etched with 35% phosphoric acid, rinsed with water, and hot air dried.
Whole exome sequencing
Genomic DNA was extracted from the saliva according to the prepIT® L2P protocol for the purification of genomic DNA from the Oragene® collecting kit (DNA Genotek Inc., Ottawa, ON, Canada). DNA samples of Patients 1 and 2 and four unaffected members of the family were sequenced using whole exome sequencing (WES) protocol to detect variants from protein-coding genes.
Exon capture was performed using the SureSelect target Enrichment system (Marcogen Inc., Seoul, Korea). Pair-end sequencing was then performed on a HiSeq 2000/2500 sequencing machine. A Burrows–Wheeler Alignment tool (BWA-0.7.12) was used to align the 100 bp reads from the sequencer to the human reference genome (hg19 from UCSC, GRCh37 from NCBI). Single-nucleotide variants (SNVs) and small INDEL variants were identified by Picard (picard-tool-1.130), Genome analysis toolkit (GATKv3.4.0), and SnpEff (SnpEff_v4.1 g). Mutation discovery was performed, using variants called from WES of six family members (two affected and four unaffected members) (Supplemental Figure S2).
Mutation analysis
Bidirectional direct sequencing (Functional Biosciences, Madison, Wisconsin, USA) and Sequencher 4.8 Sequence analysis software (Genecodes, Ann Arbor, Michigan, USA) were used to analyze the presence of variants and co-segregation between genotype and phenotype within the family. PolyPhen-2, SIFT, and mutation taster software were used to predict functional effects of the identified variants.
RESULTS
Scanning electron micrography
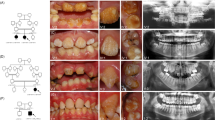
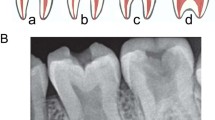
SEM images of the teeth with DI of Patient 2 at low power showed a large number of randomly scattered ectopic mineralizations on the dentinal walls of pulp chambers and root canal spaces (Figs. 6b, e, 7c, d, and 8a). At higher-power, SEM images suggested that these ectopic mineralization sites had a pattern of bush-like-mineral deposits growing outward from a focal point (Fig. 8b–f). Behind this pattern, in the background, there were more individual, plate-like deposits. It is assumed that these bush-like and plate-like deposits were minerals with their crystals larger than those of hydroxyapatite (Fig. 8c). Newly laid down dentin appeared to deposit on the bush-like mineral deposits (Fig. 8d, e). Attachment lines between the mineral deposits and dentinal wall were observed (Fig. 8b, f).

Comparison between a, d Normal tooth, b, e DI tooth of Patient 2, and c, f DI tooth of an OI patient. a Normal dentin appears smooth. Two small ectopic mineralizations are observed. b DI tooth of Patient 2. Ectopic mineralizations appear as large bush-like mineral deposits on the dentinal wall of the root canal. c DI tooth of an OI patient. Irregular surface of dentin. d–f DEJ areas of d Normal tooth. Normal dentin appears smooth. e DI tooth of Patient 2. Numerous areas of ectopic mineralizations of dentin. f DI tooth of an OI patient. Small areas of ectopic mineralizations in dentin

CDJ areas of a Normal tooth. b DI tooth of other OI patient. c, d DI tooth of Patient 2. Note numerous ectopic mineralizations on the dentin and cemental surfaces of Patient 2

Patient 2. SEM. Ectopic mineralizations of dentin surface of DI teeth of Patient 2. a–d Coronal dentin. e, f Radicular dentin. a Numerous ares of ectopic mineralizations. b Dentinal surface of pulp chamber. Attachment lines between the mineral deposits and dentinal wall (arrows). c, e Close-up view of bush-like mineral deposits. Ectopic mineralizations show well-organized pattern of bush-like mineral deposits growing outward from a focal point. The morphology of the crystals of the bush-like mineral deposits in (c) and (e) are different. d, e Predentin is deposited on the bush-like mineral deposits. f Radicular dentin. Mineral deposits appear round and square. Attachment lines between the mineral deposits and dentinal wall (arrow)
SEMs at low power of the cemento-dentinal junction (CDJ) of the teeth with DI of Patient 2 showed multiple areas of bush-like crystal projections (Fig. 9a). The close-up view of these bush-like mineral deposits at the CDJ showed long projecting crystals, similar to “French-fries” (Fig. 9b). Numerous areas of mineral deposits were scattered in the dentin and cementum surrounding the CDJ (Fig. 9a, c). Dentinal tubules were not observed. There were attachment lines joining the bush-like mineral deposits with the cemental surfaces (Fig. 9d). The sizes of the crystals appeared variable (Fig. 9e, f).

Patient 2. SEM. Ectopic mineralizations on cemental surfaces of DI teeth of Patient 2. a CDJ area. Scattered areas of mineral deposits in dentin and cementum. CDJ is filled with crystal projections. b Close-up view of CDJ. Note extension of well-organized French-fries-like crystal projections. c, d Mineral deposits on a cementum surface. Attachment lines between mineral deposits and cemental surface (arrow). e–f Mineral deposits on a cemental surface at the apical area of the incisor. Note variable sizes of crystals
SEM images of normal teeth at low power showed few ectopic mineralizations scattered on the dentinal walls of pulp chambers and the root canals (Fig. 6a, d). Their crystal patterns appeared variable, unlike those of Patient 2. Interestingly SEM images of incisors with DI of another OI patient showed irregular surfaces of dentin and areas of ectopic mineralizations near DEJ (Fig. 6c, f).
WES and mutation analysis
WES and validation by bidirectional Sanger sequencing confirmed the heterozygous base substitution c.1451G>A mutation (NM_000089.3; rs66671033) in the DNA from both affected father and daughter, but not in the four unaffected family members (Supplemental Figure S1). The father appears to have a de novo mutation; however, a paternity test was not performed. This heterozygous base substitution appears to co-segregate with the OI phenotype in this family (Supplemental Figure S2). The variant was also predicted using in silico functional annotation software to be amino acid substitution p.Gly484Glu (NP_000080.2). In particular, Mutation Taster, PolyPhen-2, and SIFT predicted this variant as 'disease causing' (0.999), 'probably damaging' (1.000), and damaging (0.000), respectively. Furthermore, this variant was not found in 100 Exome data of our normal controls or in the Exome data from the Exome Aggregation Consortium (ExAC) browser.
Discussion
We report a father and his daughter who were both affected with OI type I. Both individuals carried a heterozygous base substitution c.1451G>A mutation in COL1A2. The c.1451G>A heterozygous base substitution is predicted to cause the amino acid substitution p.Gly484Glu, which resides in the helical domain of COL1A2. The residue Gly484 is conserved in dog, rabbit, cattle, house mouse, and zebra fish (Supplemental Figure S4). The glycine substitution in the helical domain commonly causes OI. The glycine amino acid, the smallest amino acid, is crucial in every third position of the helical chain because it is the only residue small enough to occupy the sterically restricted inner aspect of the triple-stranded helical rod. Thus, glycine substitution generally causes disruption of helix folding and propagation, prolonging the access time for modification of enzymes, leading to the exposure all three chains to excess hydroxylation and glycosylation [3,4,5]. The p.Gly484Glu mutation in COL1A2 has been reported in a patient with mild OI in a supplemental table of a report [6], supporting this mutation being pathogenic. Unfortunately, the clinical description of that patient was not available. We cannot explain how a COL1A2 mutation led to the dense mandibular bone of Patient 1, while all other bones were osteopenic.
The daughter had generalized osteopenia. The father had mild osteopenia with low bone mass, as shown by dual-energy X-ray absorptiometry. It is interesting to note that Patient 1, who had OI, had no history of low-trauma bone fractures. We cannot rationalize why he did not have brittle bones. Phenotypic variability of patients affected with OI is well known. However, there are no the scientific reasons to explain the phenomenon. It appears that bone fracture needs other contributing factors besides abnormal collagens. The fractures of bones in patients affected with autosomal recessive infantile osteopetrosis supports this notion [7]. The findings from our patients suggest that this particular mutation is associated with a mild phenotype and intra-familial variability, albeit causing structural defects of collagens. Even though our patients seem to have structural defects in the helical domain of the collagens, both seem to have mild phenotypes [6].
The dentin in different teeth of Patient 2 appeared to have different patterns of ectopic mineralization. Very few dentinal tubules were observed throughout the coronal and radicular dentin, suggesting that the regulation of dentin patterning depends on the presence of normal collagen fibers. Interestingly the much milder ectopic mineralization was also detected in the dentin of another patient with OI. Ectopic mineralization in dentin and cementum of teeth affected with DI has never been previously reported. However, its presence was not a surprise because collagen type I is a major component of both dentin and cementum. Ectopic mineralizations might be the result of abnormal collagen formation because copolymerization of normal and abnormal collagens in vitro has been demonstrated to cause a distortion of normal fibril morphology, delayed fibril formation, decreased net amount of collagen incorporated into fibril, and fractal-like structures [8]. The ectopic mineralization in dentin and cementum comprised variable structures of crystals. There were attachment lines joining the bush-like mineral deposits with the dentin and cementum. The crystals of the mineral deposits in dentin and cementum appeared larger than the crystals of hydroxyapatites. The “French-fries-appearance” of the crystals at the CDJ and abnormal cementum have never been reported to be associated with DI (hereditary opalescent dentin), either isolated or OI-associated. These ultrastructural abnormalities of dentin and cementum have not been previously reported in teeth with DI [9,10,11,12] or dentin dysplasia [13] and they might be responsible for the integrity of the dentition.
Conclusions
We report a father and his daughter, both affected with OI and DI. Both were heterozygous for a COL1A2 mutation. The father had very mild OI and was a Thai boxer. Ectopic mineralizations found in dentin, cementum, and the bush-like mineral deposits at the CDJ with long projecting crystals, similar to “French-fries” at the CDJ appear to be newly recognized dental manifestations of DI. Ectopic mineralizations in dentin were also found in another patient with OI. It is hypothesized that they were caused by abnormal collagens as a result of a mutation that affected the helical domain of COL1A2. This is the first report of abnormal cementum in teeth with DI.
References
Prockop DJ, Kivirikk KI. Heritable diseases of collagen. N Engl J Med. 1984;311:376–86.
Kuivaniemi H, Tromp G, Prockop DJ. Mutations in collagen genes: causes of rare and some common diseases in humans. FASEB J. 1991;5:2052–60.
Chessler SD, Byers PH. Defective folding and stable association with protein disulfide isomerase/prolyl hydroxylase of type I procollagen with a deletion in the pro alpha 2(I) chain that preserves the Gly-X-Y repeat pattern. J Biol Chem. 1992;267:7751–7.
Engel J, Prockop DJ. The zipper-like folding of collagen triple helices and the effects of mutations that disrupt the zipper. Annu Rev Biophys Biophys Chem. 1991;20:137–52.
Forlino A, Marini JC. Osteogenesis imperfecta: prospects for molecular therapeutics. Mol Genet Metab. 2000;71:225–32.
Marini JC, Forlino A, Cabral WA, Barnes AM, San Antonio JD, Milgrom S. Consortium for osteogenesis imperfecta mutations in the helical domain of type I collagen: regions rich in lethal mutations align with collagen binding sites for integrins and proteoglycans. Hum Mutat. 2007;28:209–21.
Kantaputra PN, Thawanaphong S, Issarangporn W, Klangsinsirikul P, Ohazama A, Sharpe P. Long-term survival in infantile malignant autosomal recessive osteopetrosis secondary to homozygous p.Arg526Gln mutation in CLCN7. Am J Med Genet Part A. 2012;158A:909–16.
Kadler KE, Torre-Blanco A, Adachi E, Vogel BE, Hojima Y, Prockop DJ. A type I collagen with substitution of a cysteine for glycine-748 in the alpha 1(I) chain copolymerizes with normal type I collagen and can generate fractal like structures. Biochemistry. 1991;30:5081–8.
Wright JT, Gantt DG. The ultrastructure of the dental tissues in dentinogenesis imperfecta in man. Arch Oral Biol. 1985;30:201–6.
Lindau BM, Dietz W, Hoyer I, Lundgren T, Storhaug K, Norén JG. Morphology of dental enamel and dentine-enamel junction in osteogenesis imperfecta. Int J Paediatr Dent. 1999;9:13–21.
Gallusi G, Libonati A, Campanella V. SEM-morphology in dentinogenesis imperfecta type II: microscopic anatomy and efficacy of a dentine bonding system. Eur J Paediatr Dent. 2006;7:9–17.
Wieczorek A, Loster J. Dentinogenesis imperfecta type II: ultrastructure of teeth in sagittal sections. Folia Histochem Cytobiol. 2013;51:244–7.
Melnick M, Levin LS, Brady J. Dentin dysplasia type I: a scanning electron microscopic analysis of the primary dentition. Oral Surg Oral Med Oral Pathol. 1980;50:335–40.
Acknowledgements
We thank the patients and their family for their kind cooperation and for allowing us to use their medical and dental information for research. This work was supported by The Center of Excellence in Medical Genetics Research, Chiang Mai University; the Thailand Research Fund (TRF grant no. BRG5800013 to P.K. and RSA5860081 to S.T.); The Dental Association of Thailand; and The Faculty of Dentistry, Chiang Mai University. We also thank Dr. M. Kevin O Carroll, Professor Emeritus of the University of Mississippi School of Dentistry, USA and Faculty Consultant at Chiang Mai University Faculty of Dentistry, Thailand, for his assistance in the preparation of the manuscript. All authors declare no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Kantaputra, P.N., Sirirungruangsarn, Y., Intachai, W. et al. Osteogenesis imperfecta with ectopic mineralizations in dentin and cementum and a COL1A2 mutation . J Hum Genet 63, 811–820 (2018). https://doi.org/10.1038/s10038-018-0448-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s10038-018-0448-5
This article is cited by
-
Identification of a de novo fetal variant in osteogenesis imperfecta by targeted sequencing-based noninvasive prenatal testing
Journal of Human Genetics (2018)
