
Abstract
Gaucher disease (GD) is an autosomal recessive lysosomal storage disorder caused by the deficiency of glucocerebrosidase enzyme activity. Clinical phenotypes of GD are categorized into three groups: (i) non-neuronopathic GD (type 1), (ii) acute neuronopathic GD (type 2) and (iii) subacute neuronopathic GD (type 3). The high-risk screening of neuronopathic GD has been performed using an enzymatic assay on the dried blood spot (DBS) samples. We enrolled a total of 102 individuals (47 females, 55 males; 0–57 years old; median age 10.5 years) with various neurological symptoms. We detected two patients with very low enzyme activity and they were diagnosed with the disease by using glucocerebrosidase gene analysis. Patient 1 was found to be compound heterozygous for the p.R159W/p.R170C locus and patient 2 was found to harbor two mutations at the IVS7+1G>T (c.999+1G>T) and p.L483P sites. This simple screening protocol using DBS samples is useful for early diagnosis of GD in high-risk and underdiagnosed patients suffering from various neurological symptoms.
Similar content being viewed by others

Introduction
Gaucher disease (GD) is an autosomal recessively inherited disorder caused by mutations in the gene glucocerebrosidase (GBA; MIM number 606463) that codes for the enzyme glucocerebrosidase (GCase; EC 3.2.1.45). The deficiency of GCase results in the accumulation of glucosylceramide in lysosomes of cells in the monocyte/macrophage system, leading to a variety of systemic manifestations including hepatosplenomegaly, hematological defects, bone disease, and CNS disease [1].
Depending on the clinical features and its progression, GD has been traditionally classified into three clinical subtypes based on the absence (type 1; non-neuronopathic GD; MIM number 230800) or presence (type 2; acute neuronopathic GD; MIM number 230900 and type 3; subacute neuronopathic GD; MIM number 231000) of neurological symptoms. The prevalence of each type of GD varies with respect to the region and ethnicity. The predominant type of GD in Europe and the United States of America is type 1 (>90%) [2]. Conversely, the neurological types (type 2 and type 3) are more common (24% and 34%, respectively) in Japan [3]. As GD is rare and patients may show heterogeneous clinical symptoms, many patients are either misdiagnosed or remain undiagnosed. Recently, high-risk screening of GD patients with hepatosplenomegaly has been reported, however, high-risk screening for neuronopathic GD has not been previously investigated [4].
The screening of high-risk populations suffering from neurological symptoms could increase the diagnostic rate of GD and application of the therapeutic intervention necessary to prevent serious complications. Currently, the screening of GD is performed by measuring the GCase activity in peripheral blood leukocytes. The evaluation of dried blood spot (DBS) samples offers easy and more efficient screening.
This is an interim report of our high-risk screening program of GD. The successful detection of two patients with GD among 102 patients with various neurological symptoms emphasizes the necessity for further discussion regarding the potential of this high-risk screening method.
Materials and methods
Experimental design
During the period between April 2016 and August 2017, we analyzed DBS samples obtained from 102 patients who suffered from clinical symptoms including seizures, involuntary movements, oculomotor abnormalities, and other neurological symptoms, which might be suggestive of neuronopathic GD. All these patients had no definitive diagnosis of their respective disease. The group of patients comprised of 47 females and 55 males aged from 0 to 57 years old.
Preparation of DBS samples
All the DBS samples were prepared from peripheral blood. Blood droplets were spotted onto filter paper (Toyo Roshi Kaisha Ltd., Tokyo, Japan), were allowed to dry for ~4 h at room temperature and were sent through mail (within 1 week of preparation) to Kumamoto University for analysis. The DBS samples were stored at −20 °C until use.
GCase assay of DBS samples
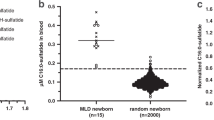
A high-throughput and reliable assay for the GCase activity was developed and it was proved to be clinically beneficial. The screening protocol for GCase assay was performed on DBS samples using the method described by Chamoles et al. [5], with a minor modification. Briefly, a 3.2-mm-diameter disk was punched from each DBS filter paper sample, and the enzyme GCase was extracted into 100 μL of extraction buffer (0.1% TritonX-100, 5 mM MgCl2, 0.5 mM dithiothreitol and 0.05% NaN3 in 25 mM citric acid–potassium phosphate buffer) (pH 6.0). The DBS sample extract (20 μL) was transferred into a black 96-well assay plate. The substrate solution (40 μL) that comprises of 3 mM 4-methylumbelliferyl-β-d-glucopyranoside (Sigma-Aldrich Corp., St. Louis, USA) and 0.3% sodium taurodeoxycholate in 100 mM citrate phosphate buffer (pH 5.0) was added to each well, and the reaction mixture was incubated for 3 h at 37 °C. In order to stop the reaction, 200 μL of 300 mM glycine-NaOH buffer (pH 10.6) was added to each well. A fluorometer was used to analyze the assay plates at 370-nm excitation and 465-nm emission wavelengths. Molar product quantities in the assay wells were calculated by linear regression from the standard curve. The enzyme activity was expressed as picomoles of 4-methylumbelliferyl-β-d-glucopyranoside released per hour per disk (pmol/h/disk). To validate our results, DBS samples were obtained from 318 healthy individuals and five previously diagnosed GD patients. Among the normal healthy control group, the GCase enzyme median activity ranged from 8.5 to 62.5 pmol/h/disk. The established cutoff values were the 5.0 pmol/h/disk (25% of the normal median activity) and 3.5 pmol/h/disk (15% of the normal median activity). The median enzyme activity ranged from 0.0 to 0.8 pmol/h/disk in five confirmed patients with GD.
Molecular analysis of GBA
The analysis method for GBA was performed by using long-range PCR and next-generation sequencing technology that had been previously reported in the corresponding reference [6]. Genomic DNA was extracted from peripheral blood using a Puregene Blood Core Kit B (Qiagen GmbH, Hilden, Germany). GBA and its flanking regions were amplified using PCR and sequenced using a MiSeq sequencer (Illumina, San Diego, CA, USA). Sequence data analysis, mapping and variant calling were subsequently processed using MiSeq Reporter v2 (Illumina). The region from 155,203,938 to 155,217,562 of the genome sequence of chromosome 1 (NC_000001.10) was used as the reference sequence.
Ethics
This study was approved by the Ethics Committee of Kumamoto University (no. 1575) and was conducted in accordance with the Declaration of Helsinki of 1975.
Results
Characteristics of patients
In this study, we enrolled 102 individuals (47 women and 55 men; range of age: 0–57 years old; median age: 10.5 years old). The inclusion criterion was the manifestation of any neurological symptom in patients. The patient group included 87 patients (85%) with seizure among whom 41 (40%) patients were affected solely by seizure and 46 (45%) patients were affected by seizure as well as another neurological symptom. Among all the patients, 21 patients (21%) had involuntary movements (19 myoclonus and two dystonia), eight patients (8%) had oculomotor abnormalities, and 64 (63%) had intellectual disabilities. Other neurological symptoms were observed in 19 patients that include hypertonia (eight patients, 8%), developmental delay (four patients, 4%), hypotonia (three patients, 3%), swallowing difficulties (two patients, 2%), and ataxia (two patients, 2%). Magnetic resonance imaging of the brain and electroencephalography showed abnormal results for 24 patients (24%) and 84 patients (82%), respectively. Organomegaly, anemia (<12.0 g/dL), and thrombocytopenia (<150 × 109/L) were observed in eight (8%), 19 (19%), and 20 (20%) patients, respectively (Table 1).
GCase assay of DBS samples
As shown in Fig. 1, GCase activity values <5.0 pmol/h/disk were observed for 6 out of 102 patients. So, we repeated GCase assay of the DBS samples acquired for the second time from these six patients. Then, GCase activity values <3.5 pmol/h/disk were observed for two out of six patients in the GCase assay repeated for the second time. Further, samples from these two patients were analyzed for mutations in GBA.

Results of beta-glucosidase assay in DBS and GBA mutation analysis
In the first GCase assay using DBS samples, the enzyme activity ranged from 0.0 to 47.3 pmol/h/disk, while in the second GCase assay using reacquired DBS samples, the enzyme activity ranged from 0.0 to 49.7 pmol/h/disk. The intra- and inter-assay coefficients of variation (CV) for GCase activity were in the range of 7.03–25.66%.
Molecular analysis of GBA
Patient 1 carried two compound heterozygous mutations: (i) c.475C>T/p.R159W acquired from the maternal allele and (ii) c.508C>T/p.R170C acquired from the paternal allele. These variants were previously reported as pathogenic mutations responsible for type 2 GD. Patient 2 carried two mutations: (i) c.999+1G>T/IVS7+1G>T and (ii) c.1448T>C/p.L483P. The IVS7+1G>T mutation is a novel variant and causes intronic base substitution at the donor splice site of exon 7 and intron 7. L483P is a common missense mutation present in several ethnic populations, including the Japanese, and its homozygosity may be associated with neurological manifestations.
Case study
Patient 1 was the first-born baby of nonconsanguineous Japanese parents. The female infant weighed 2504 g at birth and was delivered at a gestational age of 37 weeks and 5 days. Delivery was by cesarean section because of hydrops fetalis. At birth, the baby had severe collodion skin, hepatosplenomegaly, anemia (hemoglobin 8.9 g/dL), thrombocytopenia (platelets 81 × 109/L), and an elevated ferritin level (2064 μg/mL). The Apgar score was 6 (1 min) and 9 (5 min). The infant was promptly incubated and admitted to the NICU. The infant was referred to us for diagnostic aid on day 4 after her birth. The GCase assay performed using DBS samples resulted in a reading of 0.0 pmol/h/disk (cutoff <3.5 pmol/h/disk). To confirm the diagnosis, we analyzed the GBA mutation. The results indicated a compound heterozygosity at c.475C>T/p.R159W and c.508C>T/p.R170C locus and confirmed the diagnosis of type 2 GD (perinatal–lethal type). Additionally, we performed GBA mutation analyses on each parent. Her father had a heterozygous R170C mutation. Her mother had a R159W mutation at a single allele. Patient 1 died from multiorgan failure at the age of 116 days.
Patient 2 is a male infant of a Japanese ancestry and was referred to us at the age of 4 months. He suffered from hepatosplenomegaly, anemia (hemoglobin 8.7 g/dL), mild thrombocytopenia (platelets 154 × 109/L), and high acid phosphatase activity (118.6 U/L) (normal value: 5.9–14.0 U/L). Initially, the infant manifested difficulty in swallowing, abnormal eye movements, and hypertonia at the age of 3 months. The results of auditory brain stem response and blink reflex testing indicated brain stem dysfunction. GCase activity determined using DBS samples was markedly low (0.34 pmol/h/disk). GBA mutation analyses showed two mutations, i.e., c.999+1G>T/IVS7+1G>T and c.1448T>C/p.L483P. We investigated GBA mutations of his parents through gene analyses and found that his father had IVS7+1G>T mutation at a single allele and his mother had a heterozygous L483P mutation.
As of September 2017, the infant is 13 months old and had been regularly treated using enzyme replacement therapy (ERT). Hepatosplenomegaly and thrombocytopenia conditions had markedly reduced after the treatment. His respiratory function and general condition greatly improved after receiving a tracheostomy.
Discussion
In this study, we demonstrate two points: first, we were able to identify two patients with GD among a group of 102 patients with neurological symptoms and second, these two GD patients suffered from prominent features such as the abnormal eye movements and organomegaly, whereas a mere 8% among 102 patients suffered from either but not both the features.
Neuronopathic GD is relatively common in the Japanese population. One of the reasons for this is the high incidence of the L483P genotype. This genotype is typically associated with neurological symptoms.
We confirmed the diagnosis of GD in two cases among a pool of 102 patients. GBA mutation analysis revealed a compound heterozygous mutation of R159W (c.475C to T transition) and R170C (c.508C to T transition) in patient 1. Patient 2 was a compound heterozygote of L483P (c.1448T to C transition) and c.999+1G>T (IVS7+1G>T) mutation.
The R159W mutation was first reported in two Spanish patients with type 2 GD and their mutations were identified as R159W/D448H and R159W/L483P, respectively [7]. The R170C mutation was first reported in a Canadian patient with type 2 GD whose mutation was R170C/L483P [8]. The L483P mutation was reported at a high frequency in patients with neuronopathic GD [9]. The variant IVS7+1G>T is a novel mutation. The mutated nucleotide is the first nucleotide of intron 7 and we believe that it is a splicing error. The RNA sample from patient 2 was not available; so, consequently, we used in silico analysis to evaluate our hypothesis. The results of an analysis using Human Splicing Finder ver. 3 (http://www.umd.be/HSF3/) show that the mutation alters the wild-type donor site and probably affects the splicing mechanism.
Collectively, the percentage of target individuals with abnormal manifestations was low. The majority of the patients suffered from a single clinical feature with respect to the three specific clinical features of neuronopathic GD that are oculomotor abnormalities, hepatosplenomegaly and thrombocytopenia. A mere number of two and three patients suffered from two out of three and three out of three symptoms, respectively, among these specific symptoms of GD. In our study, the two GD patients suffered from two or three of these clinical manifestations (Table 1).
Patient 1 was diagnosed with perinatal–lethal-type GD. So, therapeutic possibilities were limited, however, the diagnostic confirmation provided important information to assist the infant’s parents with genetic counseling. In the case of patient 2, early diagnosis of GD led to early initiation of ERT that resulted in marked hematological improvement and improved general health in the infant. The DBS sample analysis has the potential to make the diagnostic process more cost-effective as the samples are easy to obtain and transport, which makes this diagnostic test available to populations in areas that lack the access to expensive equipment [5]. In addition, we can simultaneously conduct the routine blood sample testing, and the procedure to acquire samples is less aggressive than bone marrow aspiration [5]. Tajima et al. [3] reported that the percentage of abnormal eye movements is ~20% at the time of diagnosis and the horizontal gaze palsy is a more specific feature of GD [10]. We observed horizontal gaze palsy in patient 1. Multidirectional abnormal eye movements were observed in the case of patient 2. These observations are known to be a feature of highly severe or progressive cases [10]. The presence of hepatosplenomegaly is the most specific symptom of GD. At the time of diagnosis, hepatosplenomegaly is confirmed in ~67% of neuronopathic GD patients. The remaining patients may have other various nonspecific symptoms of GD that might result in delayed diagnosis [3].
Our study has limitations owing to the nature of sample population. The target population is susceptible to selection bias. Most of the clinical subjects had been treated in a university hospital and had various complications in addition to neurological symptoms. Moreover, most participants were treated by pediatric neurologists who are knowledgeable regarding GD and these neurologists were interested in obtaining definitive GD diagnoses. Additionally, the resultant patient population of our study was varied as the inclusion criteria were broad and as the clinical subjects referred by doctors were from a variety of specialties with respect to medical care.
We screened patients who manifested neurological symptoms by performing an enzymatic assay in elutes obtained from DBS samples on filter paper and definitively diagnosed two cases of GD. Ocular movement and liver abnormalities are characteristic symptoms of GD patients. The DBS sample testing process is inexpensive and can be included as a part of an early routine examination for patients suffering from neurological symptoms. Simple and easy inspection of GCase activity using the DBS sample test facilitates a definitive diagnosis of GD [5]. The introduction of this screening method in diagnostic tests will facilitate early identification of GD, leading to early as well as more effective treatment.
Currently, ERT is available for the initiation of treatment that is necessary to prevent the irreversible organ damage that occurs in GD as well as in the other lysosomal storage disorders, including Pompe disease, Fabry disease, and mucopolysaccharidosis. Currently, ERT and substrate reduction therapy have been developed and are being widely used for treating type 1 GD patients. Unfortunately, these therapies are ineffective for CNS symptoms. However, ERT can help in the reduction of hematological abnormalities and improve the general health of patients [11]. Ambroxol chaperone therapy for neuronopathic GD with specific GBA mutations (i.e., F213I, N188S, G193W, R120W, or G202R) has been reported to be effective [12]. Therefore, high-risk screening for the identification of patients with neuronopathic GD is helpful for rapid diagnosis in order to improve their general health condition and quality of life.
In conclusion, the high-risk screening system using DBS samples appears to be an effective method for identifying the specific disorder in the individuals with neurological symptoms caused by unidentified reasons. This simple diagnostic test is useful for specialists and general physicians to screen patients with various neurological manifestations to identify patients with neuronopathic GD.
References
Platt FM. Sphingolipid lysosomal storage disorders. Nature. 2014;510:68–75.
Charrow J, Andersson HC, Kaplan P, Kolodny EH, Mistry P, Pastores G, et al. The Gaucher registry: demographics and disease characteristics of 1698 patients with Gaucher disease. Arch Intern Med. 2000;160:2835–43.
Tajima A, Yokoi T, Ariga M, Ito T, Kaneshiro E, Eto Y, et al. Clinical and genetic study of Japanese patients with type 3 Gaucher disease. Mol Genet Metab. 2009;97:272–7.
Motta I, Filocamo M, Poggiali E, Stroppiano M, Dragani A, et al. A multicenter observational study for early diagnosis of Gaucher disease in patients with splenomegaly and/or thrombocytopenia. Eur J Haematol. 2016;96:352–9.
Chamoles NA, Blanco M, Gaggioli D, Casentini C. Gaucher and Niemann-Pick disease-enzymatic diagnosis in dried blood spots on filter paper: retrospective diagnosis in newborn-screening cards. Clin Chim Acta. 2002;317:191–7.
Yoshida S, Kido J, Matsumoto S, Momosaki K, Mitsubuchi H, Shimazu T, et al. Prenatal diagnosis of Gaucher disease using next-generation sequencing. Pediatr Int. 2016;58:946–9.
Chabas A, Cormand B, Balcells S, Gonzalez-Duarte R, Casanova C, Colomer J, et al. Neuronopathic and non-neuronopathic presentation of Gaucher disease in patients with the third most common mutation (D409H) in Spain. J Inherit Metab Dis. 1996;19:798–800.
Sinclair G, Choy FYM, Humphries L. A novel complex allele and two new point mutations in type 2 (acute neuronopathic) Gaucher disease. Blood Cells Mol Dis. 1998;24:420–7.
Tsuji S, Choudary PV, Martin BM, Stubblefield BK, Mayor JA, Barranger JA, et al. A mutation in the glucocerebrosidase gene in neuronopathic Gaucher’s disease. N Engl J Med. 1987;316:570–5.
Mignot C, Gelot A, Bessières B, Daffos F, Voyer M, Menez F, et al. Perinatal-lethal Gaucher Disease. Am J Med Genet A. 2003;120A:338–44.
El-Beshlawy A, Tylki-Szymanska A, Vellodi A, Belmatoug N, Grabowski GA, Kolodny EH, et al. Long-term hematological, visceral, and growth outcomes in children with Gaucher disease type 3 treated with imiglucerase in the International Collaborative Gaucher Group Gaucher Registry. Mol Genet Metab. 2017;120:47–56.
Narita A, Shirai K, Shinji I, Matsuda A, Ishihara A, Matsushita K, et al. Ambroxol chaperone therapy for neuronopathic Gaucher disease: a pilot study. Ann Clin Transl Neurol. 2016;3:200–15.
Acknowledgements
We thank Fumiko Nozaki, Naomi Yano, and Matsumi Harada of Kumamoto University for their excellent technical support. This study was supported by a grant from the Ministry of Health, Labor and Welfare of Japan; a grant-in-aid for scientific research from the Ministry of Education, Culture, Sports, Science and Technology and a research grant from Shire Pharmaceuticals. The institutions that provided financial support had not played a role in the data collection or analysis.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Momosaki, K., Kido, J., Matsumoto, S. et al. High-risk screening for Gaucher disease in patients with neurological symptoms. J Hum Genet 63, 717–721 (2018). https://doi.org/10.1038/s10038-018-0438-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s10038-018-0438-7
This article is cited by
-
A review of type 3 Gaucher disease: unique neurological manifestations and advances in treatment
Acta Neurologica Belgica (2024)
-
High-risk screening for Anderson–Fabry disease in patients with cardiac, renal, or neurological manifestations
Journal of Human Genetics (2019)
