
Abstract
Branchio-oto-renal (BOR) syndrome is a rare autosomal dominant disorder characterized by branchiogenic anomalies, hearing loss, and renal anomalies. The aim of this study was to reveal the clinical phenotypes and their causative genes in Japanese BOR patients. Patients clinically diagnosed with BOR syndrome were analyzed by direct sequencing, multiplex ligation-dependent probe amplification (MLPA), array-based comparative genomic hybridization (aCGH), and next-generation sequencing (NGS). We identified the causative genes in 38/51 patients from 26/36 families; EYA1 aberrations were identified in 22 families, SALL1 mutations were identified in two families, and SIX1 mutations and a 22q partial tetrasomy were identified in one family each. All patients identified with causative genes suffered from hearing loss. Second branchial arch anomalies, including a cervical fistula or cyst, preauricular pits, and renal anomalies, were frequently identified (>60%) in patients with EYA1 aberrations. Renal hypodysplasia or unknown-cause renal insufficiency was identified in more than half of patients with EYA1 aberrations. Even within the same family, renal phenotypes often varied substantially. In addition to direct sequencing, MLPA and NGS were useful for the genetic analysis of BOR patients.
Similar content being viewed by others
Introduction
Branchio-oto-renal (BOR) syndrome (BOR1 #113650, BOR2 #610896) is an autosomal dominant disorder characterized by branchiogenic anomalies, hearing loss (HL), and renal disorders including congenital anomalies of the kidney and urinary tract (CAKUT) [1]. Patients with symptoms similar to those of BOR syndrome, but without renal anomalies, are diagnosed with branchio-oto syndrome (BOS) (BOS1 #602588, BOS3 #608389). BOR syndrome and BOS are spectrum disorders; [2] therefore, they are collectively referred to as BOR syndrome here. BOR syndrome is a rare disorder; the prevalence of BOR syndrome is 1:40,000 births in European populations [3], and ~250 Japanese patients were diagnosed with BOR syndrome in 2010 [1]. The known causative genes for BOR syndrome are EYA1 (8q13.3), SIX1 (14q23.1), and SIX5 (19q13.32) [2]. In addition, pathogenic variants of SALL1 (16q12.1), the causative gene for Townes-Brocks syndrome (TBS, #107480), may display a BOR-like phenotype in affected patients [4, 5]. EYA1 is the most frequently identified gene for BOR syndrome; [2] it is a transcription factor essential for the development of the kidney, as well as the first and second branchial arches. Patients with pathogenic variants of EYA1 can present with various clinical phenotypes including and especially renal disease; renal disease is the most important prognostic factor for the patient’s quality of life. Currently, no correlations between the genotype and phenotype of BOR patients have been identified. In this study, we investigated clinical phenotypes and their causative genes in Japanese patients with BOR syndrome.
Materials and methods
Ethics statement
Genetic analyses were performed after obtaining written informed consent from all patients or their legal guardians. All procedures were reviewed and approved by the Institutional Review Board of the Kobe University School of Medicine (65 and 301), and were performed in accordance with the ethical standards established in the Declaration of Helsinki.
Patients
We analyzed patients clinically diagnosed with BOR syndrome in a Japanese population from September 2010 to September 2017. The diagnostic criteria for BOR syndrome followed those defined by Chang et al. [6]. Briefly, the major criteria were: (1) second branchial arch anomalies, including a cervical cyst (CC) and cervical fistula (CF); (2) hearing loss (HL); (3) preauricular pits (PPs); (4) auricular deformities (external ear anomalies, EEAs); and (5) renal anomalies. The minor criteria were: (1) external auditory canal anomalies; (2) middle-ear anomalies; (3) inner-ear anomalies; (4) preauricular tags (PT); and (5) facial asymmetry and palate abnormalities, among others. Patients without a family history were diagnosed with BOR syndrome if three or more of the above major criteria, or two major and at least two minor criteria, were identified. If patients had a family history of BOR syndrome, they were diagnosed with BOR syndrome if even one major symptom was identified. We excluded patients with phenotypes characteristic of other syndromes, for example, moderate to severe intellectual disability (ID) or motor developmental delay, long palpebral fissures, preaxial polydactyly, or imperforate anus. These phenotypes were more likely to be caused by CHARGE syndrome (#214800), Kabuki syndrome (#147920, #300867), or Townes-Brocks syndrome (TBS). In this study, we included patients with renal anomalies and mutations in BOR-related genes, as identified by next-generation sequencing (NGS). The characteristics of the patients are listed in Tables 1, 2, and Supplementary Table 1.
Genetic analysis
The genomic DNA of patients was extracted from peripheral blood mononuclear cells using the QuickGene whole blood kit S (Kurabo, Osaka, Japan) according to the manufacturer’s instructions. The genomic DNA of a deceased boy (the brother of SC19) was extracted from the dried umbilical cord using the QuickGene tissue kit S (Kurabo).
First, EYA1 (accession nos. NM_000503.4 and NP_000494.2), SIX1 (NM_005982.3 and NP005973.1), SALL1 (NM_001127892 and NP_001121364), and SIX5 (NM_175875.4 and NP_787071.2) were analyzed by Sanger sequencing (Supplementary Table 2). All exons were amplified by PCR. The PCR products were purified and analyzed for the direct sequencing of all exons and their boundary introns using the Dye Terminator Cycle Sequencing Kit (GE Healthcare, Little Chalfont, UK) with an ABI Prism 3130 system (Applied Biosystems, Foster City, CA, USA). For patients where a causative gene was not identified by direct sequencing, multiplex ligation-dependent probe amplification (MLPA) was performed using SALSA MLPA P153 EYA1 (for EYA1) and P180 Limb malformations-2 (for SALL1) probe mix according to the manufacturer’s instructions (MRC-Holland, Amsterdam, Netherlands).
We performed array-based comparative genomic hybridization (aCGH) or NGS on five patients using the SurePrint G3 Human CNV microarray 400 K kit or the SurePrint G3 Human CGH microarray 180 K kit (Agilent Technologies, Santa Clara, CA, USA) for aCGH, according to the manufacturer’s instructions. NGS was performed using the Illumina TruSight One (TS1) sequencing panel or HaloPlex HS on a MiSeq platform according to the manufacturer’s instructions (Illumina, San Diego, CA, USA) as previously described [7]. The gene list constructed in this study using HaloPlex HS can be found in Supplementary Table 3 (v. 2), 4 (v. 3), and 5 (v. 4). The variants were confirmed by Sanger sequencing.
Data analysis
Sanger sequencing data were analyzed using the CLC main workbench v. 6.7.1 (Qiagen, Hilden, Germany). The aCGH data were analyzed by CytoGenomics (Agilent Technologies). To analyze the data from the TS1 sequencing panel, we used VariantStudio v. 2.1.46 (Illumina) and Integrative Genomics Viewer software v. 2.3.57 (Broad Institute, Cambridge, MA, USA). The data from HaloPlex and HaloPlex HS were analyzed by SureCall v3.5 (Agilent Technologies). The reference genome used was hg19. Missense variants were evaluated using PolyPhen2 (http://genetics.bwh.harvard.edu/pph2/), SIFT (http://sift.jcvi.org/), and Mutation Taster (http://www.mutationtaster.org/). Splice site variants were evaluated using Human Splicing Finder (http://www.umd.be/HSF3/). The following databases were accessed using AlamutVisual v2.9.0 (Interactive Biosoftware, Rouen, France), the Human Gene Mutation Database (HGMD, http://www.hgmd.cf.ac.uk/ac/index.php), ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), ExAC (http://exac.broadinstitute.org/), and the Human Genetic Variation Database (HGVD, http://www.hgvd.genome.med.kyoto-u.ac.jp/) to collate healthy controls in global and Japanese populations.
Results
Patients and causative genes
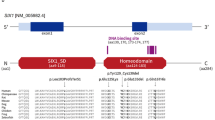
Fifty-one patients from 36 families were clinically diagnosed with BOR syndrome, and causative genes were identified in 38 patients from 26 families. Thirty-four patients from 22 families were found to contain EYA1 heterozygous mutations or partial deletions (Table 1); nonsense mutations were identified in six families (SC7, SC19, SC100, SC347, SC381, and SC462), and frameshift mutations due to the deletion or duplication of several base pairs were identified in four families (SC170, SC219, SC242, and SC289). One patient (SC7) with a nonsense mutation and another with a frameshift mutation (SC289) have been already reported [8, 9]. The same missense mutation (R440Q) was detected in three different families (SC313, SC336, and SC434). Splice site mutations were identified in three families (SC46, SC251, and SC276); exonic deletions, identified by MLPA, were identified in six families (SC4, SC5, SC6, SC11, SC112, and SC316) (Table 1, Fig. 1). One patient (SC101) had a SIX1 heterozygous missense mutation. In two patients, the causative gene was identified as SALL1; one patient (SC26) found to contain a microdeletion at 16q12, which includes SALL1, has been already reported [5], and another patient (SC334) had a SALL1 frameshift mutation caused by a single-nucleotide duplication (Table 2). Thirteen patients from 10 families with a BOR-like phenotype had no mutations in BOR-related genes (Supplementary Table 1).

Locations of EYA1 mutations identified in this study. EyaHR eyes absent homologous region, ● nonsense, ◊ splice site, ▲ missense, ■ frameshift, solid line exon deletion, broken line undetermined region
Clinical phenotypes
The phenotypes and genotypes of the patients with EYA1 aberrations are listed in Table 3. In addition, HL, which was found in all patients, PP, second branchial arch anomalies including CC or CF, and renal anomalies were highly common symptoms (>60%).
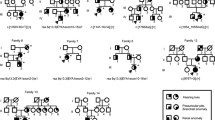
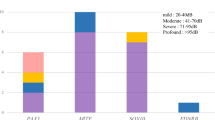
Twenty-three patients with EYA1 aberrations from 18 families were identified with renal anomalies in this study. The most frequent type of renal disease in patients with EYA1 aberrations was renal hypodysplasia (RHD), which was found in 10 patients (Fig. 2). Unknown-cause renal dysfunctions were identified in seven patients. Over 50% of the patients with EYA1 aberrations were diagnosed with RHD or unknown-cause renal insufficiency. Four patients (the mother of SC5 and SC100, SC170, and SC276) needed renal replacement therapy because of end-stage renal disease. One boy (the brother of SC19) suffered from pulmonary hypoplasia caused by bilateral renal hypoplasia, resulting in perinatal death. Even within the same families, the renal phenotypes often varied widely (Fig. 3).

Renal phenotypes in patients with EYA1 mutations. ARR abnormal renal rotation, CyK cystic kidney, HN hydronephrosis, MCDK multicystic dysplastic kidney, ReA renal aplasia, RHD renal hypodysplasia, VUR vesicoureteral reflux

Family trees showing different renal phenotypes within the same families. BOR branchio-oto-renal syndrome
Discussion
This study investigated several causative genes and clinical phenotypes relating to BOR syndrome in a Japanese population. We identified causative genes in 72.2% of patients in families clinically diagnosed with BOR syndrome. The detection rate in this study was higher than in our previous study [10]. As we mainly analyzed typical BOR syndrome patients, this may have resulted in the high detection rate. EYA1 was the most frequently identified gene in our study, consistent with previous reports on populations in Japan [11], Taiwan [12], and Western countries [10]. We identified EYA1 aberrations in 22 families. With the exception of one family (SC5 and her relatives), all EYA1 mutations were located at the 3′-end of exon 6, which includes the eyes absent homologous region, a highly conserved 271-amino acid sequence in the C-terminal region of the protein [13]. This study did not identify any correlations between the genotype and phenotype of EYA1 in BOR patients.
The following methods of genetic analysis were used for patients diagnosed with BOR syndrome: direct sequencing for 15 families, MLPA for six families, NGS for three families, and aCGH for two families. We subsequently performed MLPA analysis for undiagnosed patients with EYA1 mutations through direct sequencing. We have previously reported another Japanese patient with a LINE1 insertion in EYA1 who was diagnosed through MLPA [14]. MLPA analysis is therefore useful for the diagnosis of BOR syndrome. We identified causative genes in three BOR patients (SC101, SC170, and SC334) by NGS. A boy (SC170) in which an EYA1 aberration was identified through NGS using TS1 had severe renal insufficiency, HL, branchial anomaly, and mild ID. He also had a SIX2 missense mutation (NM_016932.4: c.390C>G, p.Cys130Trp). The patient had no family history of BOR syndrome. The SIX2 mutation was inherited from a healthy mother, but the EYA1 variant was de novo. SIX2 is an important protein for mammalian renal development, and previous reports have identified patients with renal hypoplasia and mutations in SIX2 [15, 16]. Xu et al. reported that Six2 interacts with Eya1 and Myc for regulation of nephron progenitor cells in mice [17]. In patient SC170, the SIX2 variant may have contributed to his severe renal disease, although his mother had no renal diseases. One patient (SC101) was identified with a SIX1 aberration by NGS. This variant (NM_005982.3: c.519G>C, p.Lys173Asn) has not been reported in mutational or healthy control databases. In general, BOR patients with SIX1 mutations have a lower incidence of renal disease [3]. This patient did not suffer from any renal diseases, consistent with previous reports.
SALL1 is a transcription factor that is essential for renal development [18]. Two patients were identified with SALL1 aberrations in this study. One male patient (SC334) with a SALL1 frameshift mutation was compatible with the BOR phenotype, but not TBS; another patient (SC26) had a 16q12 microdeletion that included SALL1 [5]. Neither patient presented with preaxial polydactyly or imperforate anus, which are characteristic features of TBS. It is an extremely rare disease; the prevalence of TBS is estimated to be 1:250,000 [19]. However, Engels et al. reported one BOR patient with a SALL1 frameshift mutation; [4] therefore, it is essential to analyze SALL1 in BOR patients without any EYA1 or SIX1 aberrations.
Renal anomalies are the most important complication for the prognosis of patients with BOR, because renal insufficiency may cause severe anemia, bone diseases, stunted growth, or cardiovascular disease. However, we did not identify a notable genotype–phenotype correlations for patients with BOR syndrome; even within the same family, it was impossible to predict any renal anomalies based on the presence or absence of EYA1 mutations. One girl (SC100) needed renal replacement therapy, although her father, who had the same EYA1 mutation, did not show any renal anomalies. One boy (the brother of SC19), who died 6 h after birth, had a healthy mother with the same EYA1 mutation. This unpredictability is important for the genetic counseling of families with a history of BOR syndrome. The reason for these differences remains unknown, and further study is required to resolve this uncertainty.
One patient (SC211) with left renal agenesis, HL, and EEA without ID met the criteria to be diagnosed with BOR syndrome. He had no mutations in EYA1, SIX1, SALL1, or SIX5, but did have a karyotype abnormality (47, XY +mar). We performed aCGH and diagnosed him with chromosome 22 partial tetrasomy (also known as cat eye syndrome or Schmid-Fraccaro syndrome, #115470). Chromosome 22 partial tetrasomy has an autosomal dominant pattern of inheritance, but there was no family history of the syndrome in this case. Symptoms included EEA with conductive HL, renal agenesis, and eye abnormalities. He had bilateral ptosis without a coloboma. IDs from this syndrome were non-existent or mild, and the intelligence of the patient was normal. Therefore, if patients with symptoms of BOR syndrome have any eye abnormalities, chromosome 22 partial tetrasomy should be considered as a potential alternative cause, and chromosomal tests including a G-banding test or aCGH should be performed to rule this out.
No major SIX5 mutations were identified in any of our study participants. Hoskins et al. [20] reported that SIX5 is a causative gene for BOR syndrome in 2007; however, no other published reports have confirmed the association of SIX5 with BOR syndrome, although a SIX5 mutation has been reported to be the cause of CAKUT in two patients [15, 21]. In a study on a Taiwanese population, no connection was found between BOR syndrome and SIX5 mutations [11]. In addition, Krug and colleagues found that a patient with BOR syndrome and a SIX5 mutation also had a mutation in EYA1 [9]. Although further investigation is necessary, SIX5 may not have a role in BOR syndrome, at least in East Asian populations. In this study, 13 patients from 10 families showed no mutations in BOR-related genes. NGS was performed on all patients, including TS1 or a comprehensive CAKUT gene analysis. Further studies using whole-exome or genome sequencing are necessary for the patients and their families. From our results and latest findings, we suggest an algorithm for precise diagnosis in BOR syndrome (Fig. 4).

Flow chart for genetic diagnosis in BOR patients
In conclusion, this was the first large-scale study to perform a genetic analysis on patients with BOR syndrome in a Japanese population. No obvious genotype–phenotype correlation was identified in this study. Renal abnormalities were especially unpredictable. We propose that MLPA analysis for EYA1 and comprehensive NGS analysis will be useful for the detection of genes related to BOR syndrome.
References
Morisada N, Nozu K, Iijima K. Branchio-oto-renal syndrome: comprehensive review based on nationwide surveillance in Japan. Pediatr Int. 2014;56:309–14.
Smith RJH. In: Branchiootorenal spectrum disorders. Pagon RA, Adam MP, Ardinger HH, (eds). GeneReviews. Seattle: University of Washington; 2015.
Fraser FC, Sproule JR, Halal F, Optiz JM. Frequency of the branchio-oto-renal (BOR) syndrome in children with profound hearing loss. Am J Med Genet A. 1980;7:341–9.
Engels S, Kohlhase J, McGaughran J. ASALL1 mutation causes a branchio-oto-renal syndrome-like phenotype. J Med Genet. 2000;37:458–60.
Morisada N, Sekine T, Ishimori S, Tsuda M, Adachi M, Nozu K, et al. 16q12 microdeletion syndrome in two Japanese boys. Pediatr Int. 2014;56:e75–8.
Chang EH, Menezes M, Meyer NC, Cucci RA, Vervoort VS, Schwartz CE, et al. Branchio-oto-renal syndrome: the mutation spectrum in EYA1 and its phenotypic consequences. Hum Mutat. 2004;23:582–9.
Yoshikawa T, Kamei K, Nagata H, Saida K, Sato M, Ogura M, et al. Diversity of renal phenotypes in patients with WDR19 mutations: two case reports. Nephrology. 2017;22:566–71.
Ogawa A, Kitamura S, Nakayama K, Sugiyama H, Morisada N, Iijima K, et al. Right hypoplastic kidney. Kidney Int. 2012;82:1037.
Iwaki T, Wakabayashi T, Inoue A, Irie K, Fuke N, Kondo T, et al. A case of BOR syndrome complicated by cerebral cavernous malformation exhibiting novel mutation of the EYA1. Jpn. J. Pediatr. Nephrol. (in press).
Krug P, Morinière V, Marlin S, Koubi V, Gabriel HD, Colin E, et al. Mutation screening of the EYA1, SIX1, and SIX5 genes in a large cohort of patients harboring branchio-oto-renal syndrome calls into question the pathogenic role of SIX5 mutations. Hum Mutat. 2011;32:183–90.
Okada M, Fujimaru R, Morimoto N, Satomura K, Kaku Y, Tsuzuki K, et al. EYA1 and SIX1 gene mutations in Japanese patients with branchio-oto-renal (BOR) syndrome and related conditions. Pediatr Nephrol. 2006;21:475–81.
Wang SH, Wu CC, Lu YC, Lin YH, Su YN, Hwu WL, et al. Mutation screening of the EYA1, SIX1, and SIX5 genes in an East Asian cohort with branchio-oto-renal syndrome. Laryngoscope. 2012;122:1130–6.
Abdelhak S, Kalatzis V, Heilig R, Compain S, Samson D, Vincent C, et al. Clustering of mutations responsible for branchio-oto-renal (BOR) syndrome in the eyes absent homologous region (eyaHR) of EYA1. Hum Mol Genet. 1997;6:2247–55.
Morisada N, Rendtorff ND, Nozu K, Morishita T, Miyakawa T, Matsumoto T, et al. Branchio-oto-renal syndrome caused by partial EYA1 deletion due to LINE-1 insertion. Pediatr Nephrol. 2010;25:1343–8.
Weber S, Taylor JC, Winyard P, Baker KF, Sullivan-Brown J, Schild R, et al. SIX2 and BMP4 mutations associate with anomalous kidney development. J Am Soc Nephrol. 2008;19:891–903.
Hwang DY, Dworschak GC, Kohl S, Saisawat P, Vivante A, Hilger AC, et al. Mutations in 12 known dominant disease-causing genes clarify many congenital anomalies of the kidney and urinary tract. Kidney Int. 2014;85:1429–33.
Xu J, Wong EY, Cheng C, Li J, Sharkar MT, Xu CY, et al. Eya1 interacts with Six2 and Myc to regulate expansion of the nephron progenitor pool during nephrogenesis. Dev Cell. 2014;31:434–47.
Nishinakamura R, Matsumoto Y, Nakao K, Nakamura K, Sato A, Copeland NG, et al. Murine homolog of SALL1 is essential for ureteric bud invasion in kidney development. Development. 2001;128:3105–15.
Kohlhase J. In: Townes-Brocks syndrome. Pagon RA, Adam MP, Ardinger HH, (eds). GeneReviews. Seattle: University of Washington; 2016.
Hoskins BE, Cramer CH, Silvius D, Zou D, Raymond RM, Orten DJ, et al. Transcription factor SIX5 is mutated in patients with branchio-oto-renal syndrome. Am J Hum Genet. 2007;80:800–4.
Nicolaou N, Pulit SL, Nijman IJ, Monroe GR, Feitz WF, Schreuder MF, et al. Prioritization and burden analysis of rare variants in 208 candidate genes suggest they do not play a major role in CAKUT. Kidney Int. 2016;89:476–86.
Acknowledgements
We thank all the participants and their families. We also thank Takashi Sekine (Toho University), Yoshitsugu Kaku, Pin Fee Chong (Fukuoka Children’s Hospital), Ayu Ogawa (Okayama University), Seiji Tanaka (Kurume University), Tsukasa Takemura (Kindai University), Kotaro Nomura (St Luke’s International Hospital), Midori Awazu (Keio University), Kazuaki Matsumoto (JA Toride Medical Center), Takahisa Yoshikawa (Kyoto University), Chikahiko Numakura (Yamagata University), Kei Nishiyama (Kyushu University), and Toshihiko Shirakawa (Nagasaki University) for their contributions. Furthermore, we are profoundly grateful to Tetsuko Yamanouchi and Yoshimi Nozu (Kobe University) for their technical assistance. We would like to thank Editage (www.editage.jp) for English language editing.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
This work was supported by a Health Labor Sciences Research Grant for the Research on Measures for Intractable Diseases (H21-nanchi-ippan-103 to K.I., H24-nanchi-ippan-041 to K.I., and H29-nanchi-ippan-039 to N.M.) and Japan Society for the Promotion of Science KAKENHI grant number JP15K09261 (to N.M.). The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Unzaki, A., Morisada, N., Nozu, K. et al. Clinically diverse phenotypes and genotypes of patients with branchio-oto-renal syndrome. J Hum Genet 63, 647–656 (2018). https://doi.org/10.1038/s10038-018-0429-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s10038-018-0429-8
This article is cited by
-
Novel likely pathogenic variant in the EYA1 gene causing Branchio oto renal syndrome and the exploration of pathogenic mechanisms
BMC Medical Genomics (2024)
-
Misdiagnosed Branchio-Oto-Renal syndrome presenting as proteinuria and renal insufficiency with insidious signs since early childhood: a report of three cases
BMC Nephrology (2023)
-
Hereditäre Schwerhörigkeit
HNO (2023)
-
Phenotype–genotype correlation in patients with typical and atypical branchio-oto-renal syndrome
Scientific Reports (2022)
-
The genetic basis of congenital anomalies of the kidney and urinary tract
Pediatric Nephrology (2022)
