
Abstract
Tuberous sclerosis (TS) is a rare autosomal-dominant genetic disease. TS is manifested by the development of multiple hamartomas, which affect brain, kidneys, retina, skin and other organs. This study aimed to reveal specific features of molecular epidemiology of TS in Russia. Blood DNA samples from 61 patients with definite (n = 53) or probable (n = 8) clinical diagnosis of TS were tested for mutations in TSC1 and TSC2 genes using Sanger sequencing and MLPA analysis. Five TSC1/2 mutation-negative patients were further analyzed by exome sequencing. TSC1/2 mutations were detected in 53/61 patients (87%): 39 (74%) carried mutations in the TSC2 and 14 (26%) in the TSC1. Large rearrangements (exon deletions/duplications) affected exclusively TSC2, accounting for 15% of lesions of this gene. 6/8 (75%) patients with incomplete clinical manifestation of TS carried TSC1/2 gene lesion. Overall, 96% of detected germline TSC1/2 mutations occurred de novo. Patients with no mutation identified (NMI) differed from TSC1/2 mutation carriers, being lacking cortical tubers and subependymal nodules but having higher frequencies of renal angiomyolipomas, rhabdomyomas, and lymphangioleiomyomatosis. Exome sequencing failed to identify overt disease-causing mutation candidates among NMI patients. Russian patients with TS have increased frequency of TSC2 large gene rearrangements and TSC1/2 mutations occurring de novo as compared to other studies. Patients with suspected TS diagnosis but NMI status may represent a distinct disease entity.
Similar content being viewed by others
Introduction
Tuberous sclerosis (TS) is a rare autosomal-dominant genetic disorder affecting the skin, brain, heart, kidneys, lungs, eyes, and other organs [1]. The first TS locus was mapped to the distal long arm of chromosome 9 due to co-inheritance of this disease with ABO blood groups in some of the analyzed families [2]. This led to subsequent identification of the corresponding gene, TSC1, located on 9q34 and coding for hamartin [3]. The success in the discovery of the second TS locus was attributed to the emphasis on the specific feature of TS, i.e., presence of kidney cysts. While studying TSC1-negative TS pedigrees, Kandt et al. [4]. intentionally focused on the polymorphic markers located in the vicinity to the polycystic kidney disease gene (PKD1), and mapped the TSC2 to chromosome 16p13.3. TSC2 gene is considerably larger than TSC1 and codes for tuberin [5, 6].
Both TSC1 and TSC2 act as negative regulators of mTOR signaling pathway. Their inactivation results in the appearance of multiple tumor-like lesions, hamartomas. In addition to acting as tumor suppressors, TSC1 and TSC2 are likely to play a role in a number of other biological processes, e.g., in the functioning of nervous system. TS manifestations are highly variable, being dependent on the patient age and gender, type of the TSC1/2 gene mutation as well as other, yet unrecognized factors. Most of currently diagnosed TS patients have a relatively severe disease presentation, which significantly alters their well-being. For this reason, subjects with TS have reduced chances to have a family, and therefore only less than a third of TS cases are caused by vertical transmission of the TSC1/2 germline mutation [7]. The majority of TS instances are sporadic and attributed to de novo defect in the TSC1 or TSC2 genes. In addition, TSC1/2 mutation mosaicism contributes to a share of TS morbidity [1, 7,8,9].
Clinical diagnosis of TS is based on the evaluation of so-called “major” and “minor” TS features. Major features include characteristic abnormalities of the skin and oral cavity (hypomelanotic macules, angiofibromas, ungual fibromas, shagreen patch), brain (cortical dysplasias, subependymal nodules, subependymal giant cell astrocytoma), heart (cardiac rhabdomyoma), eyes (multiple retinal hamartomas), lungs (lymphangioleiomyomatosis), and kidneys (angiomyolipomas). Confetti skin lesions, dental enamel pits, intraoral fibromas, multiple renal cysts, retinal achromic patches, and non-renal hamartomas are classified as minor TS features. Clinical TS diagnosis is considered definite if a patient has two major features of TS or combination of 1 major feature with at least two minor features. Subjects with only one major TS feature or two and more minor features belong to the category of possible TS diagnosis. Patients with genetic diagnosis of TS, i.e., presence of clearly pathogenic mutation in TSC1 or TSC2 gene, are classified as TS cases irrespectively of the clinical disease manifestation [10].
TS molecular genetics is relatively well described, with hundreds of patients already subjected to comprehensive TSC1/2 gene testing. However, the geography of TS genetic studies is limited to a number of countries, including USA, Western Europe (UK, Netherlands, Germany), Poland, India, Taiwan, Korea, China, Japan and Australia (Supplementary Table 1). The existence of ethnic and/or country-specific variations in TS presentation is acknowledged in some studies, therefore the analysis of yet unstudied populations is justified [11]. Here we report the first Russian study describing the pattern of TSC1/2 mutations in TS patients.
Materials and methods
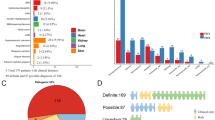
Sixty one patients with clinical signs of tuberous sclerosis (TS) were collected during years 2012–2016 in several medical facilities operating in Moscow, St. Petersburg, Ekaterinburg and Ufa; the majority of patients were from the TS registry maintained by the Center of Epileptology (N.I. Pirogov National Research Medical University, Moscow, Russia). Fifty-three (87%) of recruited patients met criteria for definite diagnosis of TS, whereas the remaining 8 (13%) were classified as possible TS cases. The mean age of patients was 8.8 years (range: 3 months – 43 years). Informed consent was obtained for all the patients or their parents or guardians prior to their inclusion in the study. The study was approved by the local Ethics Committee.
Small mutations in TSC1 and TSC2 genes were analyzed by high-resolution melting (HRM) analysis followed by Sanger sequencing of suspicious fragments. Primer sequences and PCR conditions are presented in the Supplementary Table 2. Detection of large rearrangements was carried out by multiplex ligation-dependent probe amplification (MLPA) using P124 probemix for TSC1 and P046 for TSC2; suspected TSC2 alterations were confirmed using additional P337 probemix. The exact description of the MLPA protocol is available from the web site of the kit manufacturer (MRC Holland, http://www.mlpa.com).
DNA samples from five TSC1/2 mutation-negative patients were subjected to whole-exome sequencing (WES). Exome enrichment was performed using the Nextera kit (Illumina, USA), which covers 37 Mb of coding sequences (214,405 exons; 98.3% of sequences annotated in RefSeq database). Massive parallel sequencing was carried out using an Illumina MiSeq instrument and included multiple 150-bp reads with approximately 50× coverage. The analysis of nucleotide-specific fluorescent signals was done using MiSeq Reporter software. Reads were aligned to the Human Reference Genome (version hg19) by Burrows-Wheeller Aligner. The obtained files were analyzed using GATK (Genome Analysis Tool Kit) software. The identified differences from the reference sequence were annotated using Annovar resource (www.openbioinformatics.org/annovar/).
Results
Mutational findings
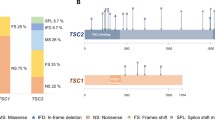
In total, TSC1/2 mutations were identified in 53/61 (87%) cases, which corresponds well to the majority of similar investigations [12,13,14,15] (Supplementary Table 1). The distribution of TSC1/2 alterations is presented in Table 1. As in other reports, TSC2 mutations were significantly more common than TSC1 gene lesions. It is essential to acknowledge a significant contribution of large rearrangements of TSC2 gene, which accounted for 15% of TSC2 gene lesions, 11% of the total number of TSC1/2 mutations and 10% of studied TS patients; these frequencies are markedly higher than in published studies [16, 17]. Combination of Sanger sequencing and MLPA allowed to reveal pathogenic mutations in 52 out of 61 patients. In addition, one previously missed TSC2 mutation was identified among five samples analyzed by WES. Re-evaluation of the original HRM protocol revealed that this was entirely a human error. Examples of WES-driven detection of TSC1/2 gene lesions overlooked by conventional genetic testing have already been described in the literature [18].
Individual clinical and genetic data for TS patients are presented in Supplementary Table 3. Several patients had hot-spot mutations in TSC2 gene. Two patients (7131, MG279) carried TSC2 c.138+1G>A allele. Another two subjects (6910, MG472) had large deletions encompassing exon 37-42 of TSC2 gene as well as a part of neighboring PKD1 gene. In addition two cases of TS (7323, MG393) were associated with TSC2 c.1832G>A (p.R611Q) mutation. All these mutations were repeatedly described in prior studies [12, 16, 19,20,21]. Noticeably, these 6 TSC2 mutations occurred de novo indicating that they represent rather the hot-spots than recurrent alleles persistent in the population; these findings are in agreement with some other reports [12], although familial cases for some of these mutations have been reported as well [13, 19, 21].
Genotype–phenotype associations
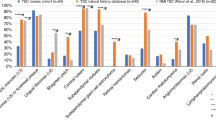
TSC1/2 gene mutations were detected in 47/53 (89%) and 6/8 (75%) patients with definite and possible diagnosis of TS, respectively. Comparison of clinical features of TS patients carrying TSC1 mutation, TSC2 gene lesion or no germline defects in these genes revealed some interesting trends (Table 2). Сases with no mutation identified (NMI) were older at diagnosis, while TSC2-associated patients had the youngest age at onset (TSC1 vs. TSC2: p = 0.004; TSC1 vs. NMI: p = 0.035; TSC2 vs. NMI: p = 0.002); these data are in good agreement with other reports [12, 22, 23]. NMI patients lacked cortical tubers and subependymal nodules thus confirming the observations of Camposano et al. [24] and Boronat et al. [25]. In addition, NMI cases were characterized by statistically lower frequency of seizures, cardiac rhabdomyomas and multiple hypomelanotic macules. These observations fit well to the results of several prior studies, which acknowledged milder phenotype of NMI patients as compared to genetically proven TS cases [12, 14, 15, 21, 24]. On the other hand, NMI patients had significantly higher frequencies of renal angiomyolipomas and lymphangioleiomyomatosis; similar trends were reported by Staley et al. [22] and Camposano et al. [24].
TSC1 mutation carriers tended to have lower male-to-female ratio and lower frequency of present or past cardiac rhabdomyomas than patients with TSC2 mutation, however these dissimilarities did not reach the threshold for statistical significance. Furthermore, only differences in the patients’ age remained significant after the adjustment for multiple comparisons; however, good agreement with published studies indicate that the observed genotype–phenotype correlations are indeed characteristic for TS disease (Supplementary Table 4).
DNA samples from the parents were available for 46 patients. Overall, 44/46 (96%) tested negative for TSC1/2 mutations identified in their children. Vertical transmission of potentially relevant mutation was documented only in 2/46 (4%) analyzed families. The TS-affected parent was detected only in one of these families (case 4972, Supplementary Table 2). In another family (MG306, Supplementary Table 2), the disease was apparently caused by TSC2 c.1865G>C (p.R622P) mutation. This mutated allele was maternally transmitted, however the mother pretended to be healthy. There are some data suggesting that c.1865G>C (p.R622P) allele [26] and some other TSC1/2 mutations [27] are associated with milder disease course. In another family (MG317), there were some clinical features of TS in the mother of the patient (facial angiofibromas and single hypomelanotic macule), however she tested negative for TSC2 exon 26-27 deletions. This can be attributed to mosaic character of TSC2 gene defect in this woman; mosaic mutations are known to be poorly detectable by conventional MLPA assay [28].
Whole-exome sequencing
Five patients were subjected to exome sequencing analysis. Two of these patients had definite diagnosis of TS (5170 и MG187, Supplementary Table 2), whereas the remaining three were adult women with renal angiomyolipomas and lymphangioleiomyomatosis (6570, MG86, MG102, Supplementary Table 2). As stated above, exome sequencing revealed previously overlooked TSC2 c.5227C>T (p.Arg1743Trp) in the patient MG187; this mutation is classified by LOVD database as probably pathogenic. We further analyzed genes, which are involved in the interaction with TSC1, TSC2, or MTOR according to BioGrid database. Twelve rare variants were identified; all were missense mutations and none occurred in more than one patient (Supplementary Table 5). MG102 carried potentially relevant CCND2 c.455C>A (p.Ala152Glu) mutation; however, this mutation was also detected in healthy mother and sister of the proband, which argued against its pathogenicity. The above variants were not functionally assessed in the present work and require further investigation to clarify their implication in TS.
Discussion
This is the first study describing Russian patients with tuberous sclerosis. The pattern of TSC1 and TSC2 mutations was generally similar to other patient series [12, 14, 21, 29]. The direct comparison of overall frequencies of TSC1/TSC2 gene defects between studies is complicated, given that the available reports utilized different techniques of mutation screening, distinct stringency of criteria for patient selection, and the clinical definitions of TS slightly evolved over the time [10, 21]. For example, Yamashita et al. [30] succeeded to detect TSC1/TSC2 only in 37% of patients analyzed; however, the median age of the patients was unusually high (19.4 years), that favored bias towards NMI cases. On another end of interstudy variations, two groups reported the detection rate of 100% [31, 32]. Although the Korean study involved only 11 patients and therefore lacked sufficient statistical power [32], the reason of the lack of NMI cases in a series of 117 Chinese patients remains obscure [31].
Our study focused mainly on patients with definite diagnosis of TS, however 8 subjects with “possible” TS were included as well; 6 (75%) of the latter were found to have TSC1/2 mutations, strongly indicating that even patients with incomplete clinical manifestation of TS should not be denied genetic testing. One could foresee that in the near future subjects with even minor symptoms of TS will be subjected to comprehensive TSC1/2 mutation testing, thanks to decreasing costs and improving accessibility of the DNA analysis. It is of interest, whether these initiatives will result in the detection of high number of virtually asymptomatic patients with genetically proven diagnosis of TS. Increased diagnostic activities towards TS patients with latent disease appearance and therefore almost normal social fitness are also likely to lead to revealing of additional instances of familial clustering of TSC1/2 mutations.
Another factor contributing to the differences in the mutation frequencies is the extent of TSC1/TSC2 gene analysis. Given that the detection of TSC1/TSC2 large rearrangements requires distinct experimental protocol and reagents, several investigators omitted the use of MLPA or similar techniques while analyzing genetic causes of TS [29, 30, 33,34,35,36]. As expected, incomplete TSC1/TSC2 gene testing resulted in somewhat lower mutation rate, with the maximal estimate approaching to 76% in the study by Hung et al. [36]. Our investigation detected large gene rearrangements in 6/53 (11%) patients with germline TSC1/TSC2 mutations. This is on the upper limit of interstudy variations, as the contribution or gross TSC1/TSC2 alterations in the total mutation spectrum ranged from 1–2% [13, 14] to 11% [37]. Overall, TSC1/2 large gene rearrangements make a noticeable contribution in TS morbidity. Therefore, the genetic analysis of TS patients should not be limited to Sanger sequencing, and MPLA testing has to be regarded as an absolutely mandatory part of examination of patients with suspected TS diagnosis.
TSC2 mutations were significantly more common in our patients than TSC1 gene lesions. Virtually all available studies on TS patients demonstrated higher frequency of TSC2 mutations as compared to TSC1 alterations, with the ratio approaching to 3–3.5:1 [12,13,14,15,16, 21, 29, 31, 38, 39]. Interestingly, Japanese TS patients appear to have an elevated share of TSC1 mutations [30, 34]; this trend does not strictly apply to other Asians, as TSC1/TSC2 mutation ratio in Chinese [31], Taiwanese [36] and Malaysian [37] patient series resembled the one in Whites, and the Korean studies produced contradictory results [32, 35]. Similarly to other reports [12,13,14, 21, 36], all TSC1 mutations in our study are truncating. In contrast, a substantial share of pathogenic TSC2 alleles is represented by amino acid substitutions (Table 1 and refs. [13, 14, 16, 19, 21]).
The proportion of familial cases in our study is clearly lower as compared to other reports (5% vs. 11–38% [12, 13, 16, 21, 30,31,32,33,34, 37, 38, 40, 41]). This can be explained by poorer social adaptation of TS patients, underdiagnosis of milder forms of TS, or low frequency of TS germline mutations in the population. Noticeable clinical differences between NMI and TSC1/2 mutation-positive patients also deserve attention (Table 2); the existence of unique characteristics of the former indicates that NMI cases cannot be explained merely by technical failure of Sanger sequencing or MLPA. It is appealing to speculate that patients with NMI represent a distinct disease, which shows phenotypic overlap with TS but has different causes. It is important to realize that the age of patients may compromise the comparison of the NMI and mutation-positive patients. For example, cardiac rhabdomyomas in TS patients tend to resolve with time [42], whereas renal angiomyolipomas and lung lymphangioleiomyomatosis often manifest later than other TS symptoms [10]. Our study is retrospective by design, therefore many of included patients could have cardiac rhabdomyomas in the past, but returned to the normal heart status by the time of TS diagnosis. Similarly, many of included patients lack kidney and lung manifestations at present, but are likely to develop them in the future. NMI patients are evidently older than the subjects with proven genetic diagnosis of TS; it is difficult to figure out, whether a unique spectrum of clinical TS presentations in NMI cases is entirely attributed to distinct biological causes of the disease, or, at least in part, simply reflects an older age at TS diagnosis. There is a popular viewpoint among TS researchers suggesting that NMI cases cannot be explained by mutation in a novel TSC gene; instead, mosaic mutations in TSC1/2 genes are detected in a subset of NMI patients [8, 43]. This corresponds well to the results of our exome sequencing study, which did not succeed to find clearly relevant germline mutations in NMI patients. However, it is important to keep in mind that WES technology has limited ability to detect gene rearrangements and therefore may miss some protein-coding mutations.
In conclusion, our study demonstrated high rate of TSC1/2 mutations among clinically diagnosed Russian TS patients. A significant share of these mutations was represented by large gene rearrangements. The proportion of de novo TSC1/2 mutations in our patient series was strikingly higher than in published studies. TS cases with no mutation identified had milder phenotype than subjects with detected TSC1/2 gene lesion. Exome sequencing of NMI patients failed to reveal a novel TS-causing gene. Taken together, these data suggest that some of the patients with clinical diagnosis of TS but absence of germline mutations in TSC1 and TSC2 genes may represent a distinct disease entity.
References
Roach ES, Kwiatkowski DJ. Seizures in tuberous sclerosis complex: hitting the target. Lancet. 2016;388:2062–4.
Fryer AE, Chalmers A, Connor JM, Fraser I, Povey S, Yates AD, et al. Evidence that the gene for tuberous sclerosis is on chromosome 9. Lancet. 1987;1:659–61.
van Slegtenhorst M, de Hoogt R, Hermans C, Nellist M, Janssen B, Verhoef S, et al. Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science. 1997;277:805–8.
Kandt RS, Haines JL, Smith M, Northrup H, Gardner RJ, Short MP, et al. Linkage of an important gene locus for tuberous sclerosis to a chromosome 16 marker for polycystic kidney disease. Nat Genet. 1992;2:37–41.
European Chromosome 16 Tuberous Sclerosis Consortium. Identification and characterization of the tuberous sclerosis gene on chromosome 16. Cell. 1993;75:1305–15.
Wienecke R, König A, DeClue JE. Identification of tuberin, the tuberous sclerosis-2 product. Tuberin possesses specific Rap1GAP activity. J Biol Chem. 1995;270:16409–14.
Curatolo P, Moavero R, Roberto D, Graziola F. Genotype/phenotype correlations in tuberous sclerosis complex. Semin Pediatr Neurol. 2015;22:259–73.
Tyburczy ME, Dies KA, Glass J, Camposano S, Chekaluk Y, Thorner AR, et al. Mosaic and intronic mutations in TSC1/TSC2 explain the majority of TSC patients with no mutation identified by conventional testing. PLoS Genet. 2015;11:e1005637. https://doi.org/10.1371/journal.pgen.1005637
DiMario FJ Jr, Sahin M, Ebrahimi-Fakhari D. Tuberous sclerosis complex. Pediatr Clin North Am. 2015;62:633–48.
Northrup H, Krueger DA, on behalf of the International Tuberous Sclerosis Complex Consensus Group. Tuberous sclerosis complex diagnostic criteria update: Recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol. 2013;49:243–54.
Jóźwiak J, Sontowska I, Płoski R. Frequency of TSC1 and TSC2 mutations in American, British, Polish and Taiwanese populations. Mol Med Rep. 2013;8:909–13.
Dabora SL, Jozwiak S, Franz DN, Roberts PS, Nieto A, Chung J, et al. Mutational analysis in a cohort of 224 tuberous sclerosis patients indicates increased severity of TSC2, compared with TSC1, disease in multiple organs. Am J Hum Genet. 2001;68:64–80.
Jones AC, Shyamsundar MM, Thomas MW, Maynard J, Idziaszczyk S, Tomkins S, et al. Comprehensive mutation analysis of TSC1 and TSC2-and phenotypic correlations in 150 families with tuberous sclerosis. Am J Hum Genet. 1999;64:1305–15.
Au KS, Williams AT, Roach ES, Batchelor L, Sparagana SP, Delgado MR, et al. Genotype/phenotype correlation in 325 individuals referred for a diagnosis of tuberous sclerosis complex in the United States. Genet Med. 2007;9:88–100.
van Eeghen AM, Black ME, Pulsifer MB, Kwiatkowski DJ, Thiele EA. Genotype and cognitive phenotype of patients with tuberous sclerosis complex. Eur J Hum Genet. 2012;20:510–5.
Rendtorff ND, Bjerregaard B, Frödin M, Kjaergaard S, Hove H, Skovby F, et al. Analysis of 65 tuberous sclerosis complex (TSC) patients by TSC2 DGGE, TSC1/TSC2 MLPA, and TSC1 long-range PCR sequencing, and report of 28 novel mutations. Hum Mutat. 2005;26:374–83.
Kozlowski P, Roberts P, Dabora S, Franz D, Bissler J, Northrup H, et al. Identification of 54 large deletions/duplications in TSC1 and TSC2 using MLPA, and genotype–phenotype correlations. Hum Genet. 2007;121:389–400.
Qin W, Chan JA, Vinters HV, Mathern GW, Franz DN, Taillon BE, et al. Analysis of TSC cortical tubers by deep sequencing of TSC1, TSC2 and KRAS demonstrates that small second-hit mutations in these genes are rare events. Brain Pathol. 2010;20:1096–105.
Beauchamp RL, Banwell A, McNamara P, Jacobsen M, Higgins E, Northrup H, et al. Exon scanning of the entire TSC2 gene for germline mutations in 40 unrelated patients with tuberous sclerosis. Hum Mutat. 1998;12:408–16.
Kwiatkowski DJ, Palmer MR, Jozwiak S, Bissler J, Franz D, Segal S, et al. Response to everolimus is seen in TSC-associated SEGAs and angiomyolipomas independent of mutation type and site in TSC1 and TSC2. Eur J Hum Genet. 2015;23:1665–72.
Sancak O, Nellist M, Goedbloed M, Elfferich P, Wouters C, Maat-Kievit A, et al. Mutational analysis of the TSC1 and TSC2 genes in a diagnostic setting: genotype–phenotype correlations and comparison of diagnostic DNA techniques in tuberous sclerosis complex. Eur J Hum Genet. 2005;13:731–41.
Staley BA, Vail EA, Thiele EA. Tuberous sclerosis complex: diagnostic challenges, presenting symptoms, and commonly missed signs. Pediatrics. 2011;127:e117–25. https://doi.org/10.1542/peds.2010-0192
Kothare SV, Singh K, Chalifoux JR, Staley BA, Weiner HL, Menzer K, Devinsky O. Severity of manifestations in tuberous sclerosis complex in relation to genotype. Epilepsia. 2014;55:1025–9.
Camposano SE, Greenberg E, Kwiatkowski DJ, Thiele EA. Distinct clinical characteristics of tuberous sclerosis complex patients with no mutation identified. Ann Hum Genet. 2009;73:141–6.
Boronat S, Shaaya EA, Doherty CM, Caruso P, Thiele EA. Tuberous sclerosis complex without tubers and subependymal nodules: a phenotype-genotype study. Clin Genet. 2014;86:149–54.
Farach LS, Gibson WT, Sparagana SP, Nellist M, Stumpel CT, Hietala M, Friedman E, et al. TSC2 c.1864C>T variant associated with mild cases of tuberous sclerosis complex. Am J Med Genet A. 2017;173:771–5.
Jansen AC, Sancak O, D’Agostino MD, Badhwar A, Roberts P, Gobbi G, et al. Unusually mild tuberous sclerosis phenotype is associated with TSC2 R905Q mutation. Ann Neurol. 2006;60:528–39.
van Veghel-Plandsoen MM, Wouters CH, Kromosoeto JN, den Ridder-Klünnen MC, Halley DJ, van den Ouweland AM. Multiplex ligation-depending probe amplification is not suitable for detection of low-grade mosaicism. Eur J Hum Genet. 2011;19:1009–12.
Niida Y, Lawrence-Smith N, Banwell A, Hammer E, Lewis J, Beauchamp RL, et al. Analysis of both TSC1 and TSC2 for germline mutations in 126 unrelated patients with tuberous sclerosis. Hum Mutat. 1999;14:412–22.
Yamashita Y, Ono J, Okada S, Wataya-Kaneda M, Yoshikawa K, Nishizawa M, et al. Analysis of all exons of TSC1 and TSC2 genes for germline mutations in Japanese patients with tuberous sclerosis: report of 10 mutations. Am J Med Genet. 2000;90:123–6.
Yang G, Shi ZN, Meng Y, Shi XY, Pang LY, Ma SF, et al. Phenotypic and genotypic characterization of Chinese children diagnosed with tuberous sclerosis complex. Clin Genet. 2017;91:764–8.
Jang MA, Hong SB, Lee JH, Lee MH, Chung MP, Shin HJ, et al. Identification of TSC1 and TSC2 mutations in Korean patients with tuberous sclerosis complex. Pediatr Neurol. 2012;46:222–4.
Ali M, Girimaji SC, Markandaya M, Shukla AK, Sacchidanand S, Kumar A. Mutation and polymorphism analysis of TSC1 and TSC2 genes in Indian patients with tuberous sclerosis complex. Acta Neurol Scand. 2005;111:54–63.
Sasongko TH, Wataya-Kaneda M, Koterazawa K, Gunadi Yusoff S, Harahap IS, et al. Novel mutations in 21 patients with tuberous sclerosis complex and variation of tandem splice-acceptor sites in TSC1 exon 14. Kobe J Med Sci. 2008;54:E73–81.
Choi JE, Chae JH, Hwang YS, Kim KJ. Mutational analysis of TSC1 and TSC2 in Korean patients with tuberous sclerosis complex. Brain Dev. 2006;28:440–6.
Hung CC, Su YN, Chien SC, Liou HH, Chen CC, Chen PC, et al. Molecular and clinical analyses of 84 patients with tuberous sclerosis complex. BMC Med Genet. 2006;7:72.
Ismail NF, Rani AQ, Nik Abdul Malik NM, Boon Hock C, Mohd Azlan SN, Abdul Razak S, et al. Combination of multiple ligation-dependent probe amplification and Illumina MiSeq amplicon sequencing for TSC1/TSC2 gene analyses in patients with tuberous sclerosis complex. J Mol Diagn. 2017;19:265–76.
Chopra M, Lawson JA, Wilson M, Kennedy SE, Taylor P, Buckley MF, et al. An Australian tuberous sclerosis cohort: are surveillance guidelines being met? J Paediatr Child Health. 2011;47:711–6.
Samueli S, Abraham K, Dressler A, Gröppel G, Mühlebner-Fahrngruber A, Scholl T, et al. Efficacy and safety of Everolimus in children with TSC—associated epilepsy—pilot data from an open single-center prospective study. Orphanet J Rare Dis. 2016;11:145.
Langkau N, Martin N, Brandt R, Zügge K, Quast S, Wiegele G, et al. TSC1 and TSC2 mutations in tuberous sclerosis, the associated phenotypes and a model to explain observed TSC1/ TSC2 frequency ratios. Eur J Pediatr. 2002;161:393–402.
Niida Y, Wakisaka A, Tsuji T, Yamada H, Kuroda M, Mitani Y, et al. Mutational analysis of TSC1 and TSC2 in Japanese patients with tuberous sclerosis complex revealed higher incidence of TSC1 patients than previously reported. J Hum Genet. 2013;58:216–25.
Bosi G, Lintermans JP, Pellegrino PA, Svaluto-Moreolo G, Vliers A. The natural history of cardiac rhabdomyoma with and without tuberous sclerosis. Acta Paediatr. 1996;85:928–31.
Nellist M, Brouwer RW, Kockx CE, van Veghel-Plandsoen M, Withagen-Hermans C, Prins-Bakker L, et al. Targeted next generation sequencing reveals previously unidentified TSC1 and TSC2 mutations. BMC Med Genet. 2015;16:10.
Acknowledgements
This work has been supported by the Russian Foundation for Basic Research (Grant No. 15-04-06714). Exome sequencing has been supported by the Russian Scientific Fund (Grant No. 15-15-00079).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Suspitsin, E.N., Yanus, G.A., Dorofeeva, M.Y. et al. Pattern of TSC1 and TSC2 germline mutations in Russian patients with tuberous sclerosis. J Hum Genet 63, 597–604 (2018). https://doi.org/10.1038/s10038-018-0416-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s10038-018-0416-0
