
Abstract
Oligonucleotide-mediated splicing modulation is a promising therapeutic approach for Duchenne muscular dystrophy (DMD). Recently, eteplirsen, a phosphorodiamidate morpholino oligomer-based splice-switching oligonucleotide (SSO) targeting DMD exon 51, was approved by the U.S. Food and Drug Administration as the first antisense-based drug for DMD patients. For further exploring SSOs targeting other exons in the DMD gene, the efficacy of exon skipping and protein rescue with each SSO sequence needs evaluations in vitro. However, only a few immortalized muscle cell lines derived from DMD patients have been reported and are available to test the efficacy of exon skipping in vitro. To solve this problem, we generated a novel immortalized DMD muscle cell line from the human rhabdomyosarcoma (RD) cell line. We removed DMD exons 51–57 (~0.3 Mb) in the RD cell line using the CRISPR/Cas9 system. Additionally, in this DMD model cell line, we evaluated the exon 50 skipping activity of previously reported SSOs at both the mRNA and protein levels. CRISPR/Cas9-mediated gene editing of the DMD gene in the RD cell line will allow for assessment of SSOs targeting most of the rare mutations in the DMD gene.
Similar content being viewed by others

Introduction
Duchenne muscular dystrophy (DMD) is an X-linked progressive muscle disorder that occurs in 1:3500–5000 newborn boys (DMD; MIM #310200) [1]. DMD is caused by mutations in the DMD gene encoding the 427 kDa dystrophin protein, which plays a major role in membrane stability in skeletal muscle [2, 3]. The mutations in DMD gene lead to two forms of diseases depending on the translational reading frame: a severe DMD and a milder Becker muscular dystrophy (BMD; MIM #300376). The mutations in DMD patients disrupt the open reading frame of the DMD gene, leading to the loss of dystrophin protein, while BMD is caused by the frame maintaining mutations. It has been revealed that the major mutation type in DMD is deletions (one exon or larger) that cause out-of-frame splicing [4].
A new therapeutic strategy for DMD patients is exon skipping therapy, which leads to an in-frame deletion of targeted exons [5]. Since Dominski et al. revealed that splice-switching oligonucleotide (SSO) has the ability to modulate splicing, exon skipping therapy has been extensively studied [6, 7]. As a result, eteplirsen, targeting DMD exon 51, was approved by the U.S. Food and Drug Administration (FDA) as the first SSO for the treatment of DMD patients in September 2016. In addition, SSOs targeting other exons in the DMD gene have been in clinical trials (https://clinicaltrials.gov/) [8]. The demand for exon skipping therapy in DMD patients will be further expanded in the future.
Exon skipping efficacy requires the proper design of the target regions, chemistries, and length of SSOs [9,10,11]. Because it is important to perform in vitro assays for the evaluation of SSO designs, many in vitro assay systems have been reported. Among them, patient-derived myogenic cells are usually used because they can be used to detect the restoration of dystrophin expression [12,13,14,15,16]. For the future development of SSOs, it is necessary to investigate exon skipping efficiency with muscle biopsies in patients, but it is difficult to obtain patient cells that have all the various mutations in the DMD gene [17]. In addition, previous reports have noted that primary skeletal muscle cells need to be immortalized [18]. Also, recent DMD guidelines do not recommend muscle biopsy if multiplex ligation-dependent probe amplification (MLPA) analysis can reveal the mutations in the DMD gene [19]. There might soon be a lack of patient biopsies to use in drug development.
The aim of the present study was to establish a novel assay cell line for the evaluation of SSOs, and thus the DMD gene in the human rhabdomyosarcoma (RD) cell line was edited. Among the cell lines from muscle, RD cell line is one of the well-studied cell lines and has been reported to express DMD mRNA [20]. Also, it is easier to induce differentiation of RD cells. According to previous reports, only 12-O-Tetradecanoylphorbol-13-Acetate (TPA) addition is enough to induce differentiation of RD cells [21, 22]. Because of the rapid growth rate of RD cell line than the immortalized myoblast cell line, we can reduce the lot-to-lot variations between the established DMD model cells. Additionally, the RD cell line is an ethically acceptable source to establish the DMD model cell line. We induced the genetic deletion with a new targeted genome editing system known as the clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 system, based on a reported mutation pattern in the DNA variant database. With the CRISPR/Cas9 system, it is possible to perform targeted gene deletion by using two guide RNAs [23].
We further confirmed the exon skipping efficiency of SSOs with the constructed cell line. As a result, we succeeded in evaluating the efficiency of previously reported SSOs at both the mRNA and protein levels.
Material and methods
Synthesis of oligonucleotides
We used five SSOs in this study (Supplementary Table S1). 2′-O-methyl RNA was incorporated into all SSO sequences, in which the phosphodiester linkages were completely replaced by phosphorothioate linkages. All SSOs were synthesized and purified by GeneDesign Inc. (Osaka, Japan). SSOs targeting DMD exons 50 or 51 were previously reported by Aartsma-Rus et al. [12]. SSOs targeting DMD exon 58 were optimized by us. We designed three SSOs, which have different lengths (13-mer to 18-mer) according to previous reports [10]. Although the 13-mer is shorter than commonly used 2′-O-methyl RNA-based SSOs, we used it for optimization because we previously reported that a 13-mer LNA/DNA mixmer showed the higher exon skipping activity [9].
Cell culture
RD cells were purchased from the American Type Culture Collection (ATCC; CCL-136). The cell line was cultured in Dulbecco’s modified Eagle Medium (DMEM) (Nacalai Tesque, Kyoto, Japan) containing 10% fetal bovine serum (FBS) (Biowest, Nuaillé, France) with antibiotic antimycotic solution (Sigma-Aldrich, St. Louis, MO) and maintained in a 5% CO2 incubator at 37 °C.
CRISPR plasmid construction
To identify candidate guide RNA sequences that were perfectly matched to the target sequence, we used the CRISPR design tool (http://crispr.mit.edu/) launched by the Zhang lab. In this analysis, we considered the score of the results and selected three sequences for each target DMD intron (50 and 57) (Supplementary Table S2). The plasmid DNAs coding the spCas9 protein, guide RNAs, and fluorescence protein (ZsGreen/tdTomato) were generated with the Guide-itTM CRISPR/Cas9 System (Green/Red) (Clontech Laboratories, Palo Alto, CA) according to the manufacturer’s protocol. Sense and antisense DNA oligonucleotides were synthesized by Integrated DNA Technologies (Coralville, IA) (Supplementary Table S3). All constructs were verified by DNA sequencing using the Applied Biosystems 3130xl Genetic Analyzer (Thermo Fisher Scientific, Pittsburgh, PA).
Plasmid DNA transfection into the RD cell line and sorting with flow cytometry
The RD cells were seeded 48 h before transfection at a density of 6.6 × 105 cells/well in T-25 flasks. Cells were transfected with a total of 20 μg plasmid DNA using Lipofectamine 2000 (Thermo Fisher Scientific) according to the manufacturer’s protocols. Forty-eight hours after transfection, cells were washed with phosphate-buffered saline (PBS) and trypsinized with 0.05% trypsin for FACS analysis. Double positive (both ZsGreen and tdTomato expression) cells were sorted with a BD FACSAria II (BD Biosciences, San Jose, CA). After fluorescence-activated cell sorting (FACS), the cells were seeded into 24-well plates (Thermo Fisher Scientific) in DMEM containing 10% FBS with antibiotics and maintained in a 5% CO2 incubator at 37 °C. For several weeks, the cells were scaled up to T-75 flasks and then cloned by limiting dilutions.
SSO transfection into the DMD model cell line
For SSO transfection, the DMD model cells were seeded 7 days before transfection at a density of 4.0 × 105 cells/well on 12-well collagen type I-coated plates (Iwaki Technoglass, Tokyo, Japan). After 24 h, cells were differentiated by changing the medium to DMEM containing 10% FBS with antibiotics and 100 nM TPA (Cell Signaling Technology, Danvers, MA) for 6 days [21, 22, 24]. Cells were transfected with 500 nM (for RT-PCR)/100 nM (for western blotting) SSOs using Lipofectamine 2000 according to the manufacturer’s protocols in DMEM containing 2% FBS with antibiotics and 100 nM TPA.
Genomic PCR
Total genomic DNAs were isolated from the cells using the QuickGene 800 (Wako, Tokyo, Japan) and QuickGene DNA tissue kit S (KURABO, Osaka, Japan) according to the manufacturer’s instructions. Total genomic DNAs were used as a template for individual PCR reactions using specific primer sets (Supplementary Table S4), which were designed using the Primer-Blast program written by Ye et al. [25]. PCR reactions were performed using KOD FX Neo DNA polymerase (TOYOBO, Osaka, Japan), and the PCR products were analyzed on a 1.5% agarose gel stained with ethidium bromide, with specific bands purified for sequence analysis. Gene edited cells were confirmed for the sequence at the breakpoint of the deletion using the Applied Biosystems 3130xl Genetic Analyzer.
MLPA analysis
Multiplex ligation-dependent probe amplification (MLPA) analysis was performed using SALSA MLPA probemix P035-B1 DMD-2 and SALSA MLPA EK1 reagent kit—FAM-labeled PCR primer (MRC-Holland, Amsterdam, the Netherlands). The reactions were performed according to the manufacturer’s instructions. The samples were analyzed by capillary electrophoresis on the Applied Biosystems 3130xl Genetic Analyzer using GS 600 LIZ size standard as a size marker. The peaks were analyzed using the Peak Scanner Software v2 (Thermo Fisher Scientific). The MLPA fragment analysis was performed using Coffalyser.Net software (MRC-Holland).
Karyotyping analysis
Karyotyping analysis was performed according to previous report [26]. The RD cells and DMD model cells were seeded into T-25 flask, respectively, and collected when the cell density is over 60%. Images were acquired with Leica TCS SP5 and SP8 (Leica, Tokyo, Japan). The number of chromosomes was counted in 20 cells, respectively.
Off-target analysis
The potential off-target sequences of guide RNAs were determined using BLAST search and CRISPRdirect (http://crispr.dbcls.jp/). The primer sets for several genes were designed to target flanking sites of potential off-target sequence (Supplementary Table S5). The genomic PCR in the DMD model cells was performed with the KOD -Multi & Epi- (TOYOBO) according to the manufacturer’s protocol. The genomic DNA extracted from RD cells was used as a reference gene. The PCR product was analyzed to determine the mutations by Sanger sequencing using the Applied Biosystems 3130xl Genetic Analyzer.
RT-PCR analysis
Twenty-four hours after transfection, total RNA samples were isolated from the cells using the QuickGene 800, QuickGene RNA cultured cell kit S (KURABO), and RQ1 RNase-Free DNase (Promega, Madison, WI) according to the manufacturer’s instructions. Reverse transcription of the total RNA samples was performed using the Rever Tra Ace qPCR RT Master Mix (TOYOBO) according to the manufacturer’s instructions. The cDNA was used as a template for individual PCR reactions using specific primer sets (Supplementary Tables S6 and S7), which were designed using the Primer-Blast program [25]. PCR reactions were performed using KOD FX Neo DNA polymerase (TOYOBO), and the PCR products were analyzed on 1.5 or 2.0% agarose gels in Tris-Acetate-EDTA (TAE) buffer stained with ethidium bromide, with specific bands purified for sequence analysis. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as an internal control.
RT-qPCR analysis
The reporter cells previously reported by our group were transfected with the SSOs (10 nM) for 24 h [9]. The expression levels of DMD exon 58-skipped mRNA fragments were measured and normalized against the signal of RPLP2 mRNA, relative to the value in the SSO_e58_15-mer set as 1. The primers used in this analysis are shown in (Supplementary Table S8).
Western blotting analysis
Four days after transfection, total cellular proteins were extracted with T-PER Tissue Protein Extraction Reagent (Thermo Fisher Scientific) and Halt Protease Inhibitor Cocktail (Thermo Fisher Scientific) according to the manufacturer’s protocols. The concentrations of total cellular protein were determined with protein assay CBB solution (Nacalai Tesque) according to the manufacturer’s protocol. Total cellular proteins were loaded onto NuPAGE 3–8% Tris-acetate protein gels (Thermo Fisher Scientific) following the manufacturer’s protocols. The gels were placed on XCell Sure Lock Mini Cell (Thermo Fisher Scientific) and were run in NuPAGE Tris-acetate SDS running buffer (Thermo Fisher Scientific) at 150 V for 65 min. The gels were transferred onto an immobilon polyvinylidene difluoride (PVDF) membrane (Millipore, Billerica, MA) by semidry blotting at 20 V for 70 min [27]. The PVDF membrane was blocked in Blocking One (Nacalai Tesque). A rabbit polyclonal antibody against the dystrophin C terminus (1:8000) (ab15277, Abcam, Cambridge, MA), a mouse monoclonal antibody against myosin heavy chain (1:8000) (MAB4470, R&D Systems, Minneapolis, MN), and a rabbit monoclonal antibody against GAPDH (1:10,000) (ab181602, Abcam) were used as primary antibodies. The primary antibodies were diluted in Can Get Signal Immunoreaction Enhancer Solution 1 (TOYOBO). The PVDF membranes were incubated with the primary antibodies for overnight at 4 °C. Horseradish peroxidase-conjugated anti-rabbit or anti-mouse goat immunoglobulins (R&D Systems, 1:100,000) were used as secondary antibodies. The secondary antibodies were diluted in Can Get Signal Immunoreaction Enhancer Solution 2 (TOYOBO). The PVDF membranes were incubated with the secondary antibodies for 1 h at room temperature. ChemiLumiOne Kit L/super/ultra (Nacalai Tesque) was used for the proteins detection according to the manufacturer’s protocol. The blots were visualized using ImageQuant LAS 4000 (GE Healthcare, Little Chalfont, UK). The total proteins extracted from differentiated human skeletal muscle myoblasts (HSMM) were used as a marker of dystrophin protein (427 kDa). The HSMM cells in the T-75 flask were cultured in DMEM containing 10% FBS with antibiotics. When the cells reached over 80% confluent, cells were differentiated by changing the medium to DMEM containing 2% horse serum (Thermo Fisher Scientific) with antibiotics for 10 days.
Immunohistochemistry analysis
The expression of dystrophin and myosin heavy chain proteins were detected by immunohistochemistry analysis. Twenty hours before inducing differentiation, RD cells were seeded at a density of 1.2 × 106 cells/well onto the gelatin-coated cover glasses (Matsunami Glass, Osaka, Japan) in 6-well culture plate (IWAKI). The cells were incubated in DMEM containing 10% FBS with antibiotics. Twenty-four hours after seeding, the medium changed into DMEM containing 10% FBS with antibiotics and 100 nM TPA for 1, 7, and 10 days. After incubation, the cover glasses were washed twice with PBS and fixed using 4% paraformaldehyde phosphate buffer solution (Nacalai tesque) for 10 min. The cover glasses were washed three times with PBS and incubated in 0.1% Triton X-100 for 10 min. The cover glasses were blocked for 1 h in PBS containing 10% goat serum (Wako, Osaka, Japan). The cover glasses were incubated in blocking solution containing primary antibodies for overnight at 4 °C. A rabbit polyclonal antibody against the dystrophin C terminus (1:50) (ab15277, Abcam) and a mouse monoclonal antibody against myosin heavy chain (1:50) (MAB4470, R&D Systems) were used as primary antibodies. After washing three times with PBS, the cover glasses were incubated in the blocking solution for 1 h at room temperature. Both AlexaFluor 488-conjugated goat anti-rabbit IgG secondary antibody (1:1000) and AlexaFluor 633-conjugated goat anti-mouse IgG secondary antibody (1:1000) were used as secondary antibodies. After washing three times, the cover glasses were mounted on glass slides (Matsunami Glass) using SlowFade Gold Antifade Mountant with DAPI (Thermo Fisher Scientific). Images were acquired with FV3000 confocal laser scanning microscope (Olympus, Tokyo, Japan).
Results
Analysis of the expression level of dystrophin in the RD cell line
We investigated the expression of DMD mRNA in differentiated RD cells at 1, 4, 7, 10, and 13 days after differentiation (Fig. 1a and Supplementary Table S6). Under the same time course of mRNA expression assays, western blotting analysis was performed to detect dystrophin protein (Fig. 1b). The results revealed that both mRNA and protein were readily detected after 7 days, with protein sufficiently detected after 10 days. Additionally, we also performed immunohistochemistry analysis and revealed that the dystrophin proteins were well expressed in the RD cells at 7 and 10 days after inducing differentiation (Supplementary Fig. S1). Therefore, we decided to use the RD cell line for the construction of a DMD model cell line due to its expression of DMD mRNA and dystrophin protein.

The levels of DMD mRNA and dystrophin protein expression in RD cell line. a The level of DMD mRNA and b dystrophin protein expressions were detected by a RT-PCR analysis or b western blotting analysis. Total RNA and total protein samples were collected at 1, 4, 7, 10, and 13 days after differentiation with 100 nM TPA. Water: no template for PCR. HSMM (100%): 100% (weight/weight percentage) total protein extracted from differentiated human skeletal muscle myoblasts. MHC myosin heavy chain. Both RT-PCR analysis and western blotting analysis were duplicated and repeated three times to ensure results were reproducible
Construction of a DMD model cell line with the CRISPR/Cas9 system
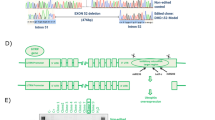
We used the Leiden muscular dystrophy pages to search for DMD mutations to construct DMD patient model cells [17]. We selected DMD patients who have one of the minor mutation patterns (patient data #0006455/#0015667/#0018293). These patients have deletion mutations of DMD exons 51–57, which can be treated with DMD exon 50 skipping therapy (Fig. 2a). However, only three patients with this mutation pattern have been reported, and thus it appears difficult to obtain biopsies.

Construction of the DMD model cell line with CRISPR/Cas9 systems. a The reported deletions in the Leiden database. The black bar indicates the deleted region of three patients (patient data #0006455/#0015667/#0018293). b Schematic representation of genetic deletion with the CRIPCR/Cas9 system. c Schematic representation and detailed target sequences of designed guide RNAs targeting DMD intron 50 and 57. The triangles indicate guide RNAs. The red triangles show that guide RNAs intron 50–3 and intron 57–1 each. The sequences highlighted in gray show the target sites of guide RNAs, and underlined sequences show the PAM sequences. d Schematic representation of plasmid DNAs. Plasmid DNAs code a Cas9 protein, guide RNA (gRNA), and fluorescence proteins (ZsGreen or tdTomato). The CMV promoter of the plasmid is bi-directional. U6: U6 promotor; gRNA: guide RNA; CMV: bi-directional CMV promotor. e The histogram of FACS sorting with fluorescence intensities of ZsGreen plotted against tdTomato. Double positive cells that express both ZsGreen and tdTomato were collected accordingly to the expression of fluorescence proteins. This histogram shows the RD cell line transfected with the guide RNA combination of “i50-3 and i57-1.” The percentage of cells that were double positive for ZsGreen and tdTomato was 3.7% in this histogram. f The efficiency of genetic deletion (% deleted) with each combination of guide RNAs was assessed by RT-PCR analysis. The sorted cells were cultured in a T-25 flask. At over 80% confluent, the cells were differentiated in medium containing 100 nM TPA. Seven days after differentiation, the levels of DMD mRNA expression were investigated by RT-PCR. Water: no template for PCR. RT-PCR analysis was duplicated and repeated three times to ensure results were reproducible (Color figure online)
To perform targeted gene deletion with the CRISPR/Cas9 system, the top three guide RNA sequences were selected according to higher-quality score outputted by the MIT CRISPR design tool (Fig. 2b, c and Supplementary Table S2). Plasmid DNAs were constructed with the Guide-itTM CRISPR/Cas9 System (Clontech Laboratories) according to the manufacturer’s protocol (Fig. 2d). The plasmid DNAs were transfected into the RD cell line, and after 2 days, the double positive cells that expressed both ZsGreen and tdTomato fluorescence proteins were collected with FACS (Fig. 2e). The sorted cells were differentiated, and then RT-PCR analyses were performed with specific primers to evaluate the efficiency of genetic deletion at the mRNA level (Fig. 2f and Supplementary Table S6). In the figure, the lower band shows the targeted genetic deletion in DMD mRNA. Based on the intensity of bands in the RT-PCR analysis, the efficiency of gene deletion was estimated. We revealed that the combination of guide RNAs “i50-3 and i57-1” was the most efficient for targeted genetic deletion. Although the combination of guide RNAs “i50-2 and i57-1” also showed the targeted genetic deletion, the efficiency was lower than guide RNA “i50-3 and i57-1.” Additionally, the other combinations did not show enough targeted genetic deletion. The DMD model cell line was acquired by limiting dilution cell culture from sorted cells with the combination of guide RNAs “i50-3 and i57-1.” We obtained 27 clones and investigated the expression of DMD mRNA extracted from 18 differentiated clones. We finally obtained a single clone that expresses the DMD exon 51–57 deleted mRNA (data not shown), and termed the cells as the DMD model cell line.
Assessment of the DMD model cell line
To assess the DMD model cell line, we performed genomic DNA analysis and evaluated the levels of DMD mRNA and dystrophin protein expressions. At first, to confirm the deletion in the DMD gene, genomic PCR was performed with specific primers (Fig. 3a and Supplementary Table S4). As we expected, genomic DNA sequencing revealed a deletion from the DMD intron 50 to intron 57 regions. In addition, we performed an MLPA analysis to confirm that the DMD model cells have a homozygous genetic deletion of DMD exon 51–57. The MLPA analysis showed that two copies of the DMD exon 51–57 were favorably deleted with the CRISPR/Cas9 system (Fig. 3b). On the other hand, the copy numbers of exons outside the genetic deletion were the same between DMD model cells and RD cells (Supplementary Table S9). We also confirmed the stability of chromosomes after CRISPR-mediated deletion by karyotyping analysis. There is no significant difference in the number of chromosomes between RD cells and DMD model cells (Supplementary Table S10). In silico analysis with the NCBI program showed that guide RNAs have the potential to have off-target effects, but genomic PCR for several genes confirmed no off-target effects (Supplementary Table S11). Next, we performed RT-PCR with specific primer sets and detected a short band in the DMD model cells (Fig. 4a and Supplementary Table S6). Sequencing of the short band showed the deletion of the exon 51–57 region in DMD mRNA (Fig.4a). Western blotting analysis showed the lack of dystrophin protein, indicating that a premature termination codon (PTC) arose at DMD exon 58 and introduced nonsense-mediated decay (NMD) (Fig. 4b).

Genomic analysis of the DMD model cell line. a Genomic DNA sequencing of the deleted region in the DMD gene. Genomic PCR was performed with specific primers. The bands before and after the CRISPR/Cas9 treatment were sequenced to confirm the original genomic sequence between two target sites of guide RNAs and genomic deletion after the treatment. b MLPA analysis of the DMD gene in RD cell line and DMD model cell line. The peak highlighted in yellow and pointed by arrows indicate the seven exons (DMD exon 51–57). RD: genomic DNA extracted from RD cell line, DMD: genomic DNA extracted from DMD model cell line (Color figure online)

Assessment of the DMD model cell line. a The expression level of DMD mRNA was assessed by RT-PCR analysis. The total RNAs were collected from the DMD model cell line at 7 days after differentiation with 100 nM TPA. RT-PCR shows the full-length (upper band: 2174 bp) and deletion (lower band: 934 bp). Water: no template for PCR. The bands were sequenced to confirm the genomic deletion at mRNA levels. b The expression level of dystrophin protein was assessed by western blotting analysis. The dystrophin protein (427 kDa) was detected only with RD cell line without gene editing. MHC myosin heavy chain. HSMM (100%): 100% (weight/weight percentage) total protein extracted from differentiated human skeletal muscle myoblasts. RD RD cell line, DMD DMD model cell line. Both RT-PCR analysis and western blotting analysis were duplicated and repeated three times to ensure results were reproducible (Color figure online)
Evaluation of 2′-O-methyl RNA SSOs with the DMD model cell line
Next, we evaluated the exon skipping efficiency of SSOs with the DMD model cell line. Fully 2′-O-methyl RNA-modified SSOs for DMD exon 50 skipping (SSO_e50) have been optimized by Aartsma-Rus et al. [12, 13]. We sought to confirm whether the DMD model cell line could be used for the evaluation of SSO_e50 at both the mRNA and protein levels. In this assay, we also used SSOs for DMD exon 51 or exon 58 skipping as negative controls (SSO_e51, SSO_e58) (Fig. 5a). The SSOs were also fully modified with 2′-O-methyl RNA. Because DMD exon 51 is deleted in the DMD model cell line, SSO_e51 will not show any effects in DMD mRNA. On the other hand, SSO_e58 can cause a frameshift of DMD mRNA, but the restoration of the dystrophin protein cannot be detected because the new exon junction between DMD exons 50–59 contains a PTC (Fig. 5a). SSO_e58 was newly designed according to our previous report (Supplementary Fig. S2 and Supplementary Table S8) [9]. As a result, although RT-PCR showed that both SSO_e50 and SSO_e58 each induced exon skipping, only SSO_e50 showed the restoration of the truncated dystrophin protein as expected (Fig. 5b, c and Supplementary Table S7). Additionally, we investigated the dose dependency of SSO_e50 with the DMD model cells (Supplementary Fig. S3). The production of DMD exon 50 skipped mRNA and shorter dystrophin proteins were promoted by SSO_e50 in a dose-dependent manner.

Evaluation of exon skipping efficiency with the DMD model cell line. a Schematic representation of exon inclusion and skipping in the DMD model cell line. The DMD model cell line expresses an out-of-frame transcript. SSO-mediated DMD exon 50 or 58 skipping result in in-frame transcripts. While exon 58 skipping introduces premature termination codon (PTC) at exon 50/59 junction leading to nonsense-mediated decay, exon 50 skipping leads to the restoration of dystrophin proteins. NMD nonsense-mediated decay. b Exon skipping efficiency of each SSO was assessed by RT-PCR. c The expression level of dystrophin protein was assessed by western blotting analysis. Western blotting analysis of restored dystrophin protein with a rabbit polyclonal antibody against dystrophin C terminus was performed at 4 days after SSO transfection into DMD model cell line. Mock: treated with Lipofectamine 2000 only; no treatment: no transfection. Water: no template for PCR. HSMM (10%): 10% (weight/weight percentage) total protein extracted from differentiated human skeletal muscle myoblasts. MHC myosin heavy chain. Both RT-PCR analysis and western blotting analysis were duplicated and repeated three times to ensure results were reproducible (Color figure online)
Discussion
In this study, we established a DMD model cell line by inducing a large deletion in the DMD gene according to the mutation database of DMD patients. We also found that the DMD model cell line enabled us to evaluate exon skipping efficiency at both the mRNA and protein levels; thus, this cell line can be a powerful tool for the confirmation of the efficacy of SSOs for DMD patients. In addition, this protocol for cell establishment enables us to establish deletion models for various genetic disorders and to provide disease cell models for both basic and clinical research.
We induced a large deletion (323 kbp) in the DMD gene by using the CRISPR/Cas9 system and established the DMD model cell line with the RD cell line. According to previous reports, combinations of two guide RNAs were able to produce targeted DMD deletions in primary skeletal muscle cells, iPS cells, or mouse muscle cells via double-stranded breaks and non-homologous end joining-mediated DNA repair [28,29,30,31]. However, the efficiency of genetic deletion seems to be low. As far as we know, one of the examples of better efficiency was reported by Ousterout et al.; the efficacy of short genetic deletion (<1 kbp) with DMD myoblast cells was less than 13.6% [28]. Because it seems to be difficult to induce larger deletions (323 kbp), we introduced a modified cell collection protocol using FACS sorting. Using two fluorescence proteins to confirm the expression of the Cas9 protein and the two guide RNAs, we succeeded in constructing the DMD model cells although it was challenging to induce a very large deletion.
The efficiency of large genetic deletion was estimated at 5% according to the number of clones obtained by limiting dilution; one out of eighteen clones has an expected homozygous deletion. Additionally, the MLPA analysis showed the similar efficiency of genetic deletion (Supplementary Table S9). Considering this, an improvement of the gene editing efficiency of the CRISPR/Cas9 system is desired in the future experiments. Among possible strategies, we can suggest four points; performing the Surveyor assays, designing as many guide RNAs as possible, using multiple in silico tools to design guide RNAs, and improving transfection efficiencies of plasmid DNAs. In this study, we designed three guide RNAs targeting DMD intron 50 and 57, respectively. As a result, we revealed that only two combinations “i50-2 and i57-1” and “i50-3 and i57-1” showed the targeted gene deletion in the evaluation of combinations of guide RNAs (Fig. 2f). This result means that only three out of six guide RNAs have the cleavage activities. In fact, the result of Surveyor assays in HEK293 cells corresponds to the result in Fig. 2f; at least the guide RNAs, i50-2, i50-3, and i57-1 showed the cleavage activities (Supplementary Fig. S4). In the future experiment, the Surveyor assays for investigating the cleavage activities of individual guide RNAs should be performed before establishing the DMD model cells. Additionally, we can suggest designing as many guide RNA as possible to pursue much higher efficiency of gene deletions. Next, using multiple in silico tools can also be suggested for efficient genetic deletion. In the study, we used the CRISPR design tool (http://crispr.mit.edu/) to design the guide RNAs. However, the in silico analysis using Cas-OFF finder (http://www.rgenome.net/cas-offinder/) revealed that the guide RNA i57-3 had the four perfectly matched off-target sites. It seems to be better using multiple in silico tools to compare the designed guide RNAs. Last, the transfection efficiencies should be improved for better efficiency of gene deletion. In the result of FACS sorting in Fig. 2e, the double positive cells were only 3.7%. For example, it might be better selecting viral transfection in the future experiment.
Our results showed that the DMD model cell line can be used to evaluate the exon skipping efficiency of SSOs. In the near future, the application of SSOs for various mutations in the DMD gene is expected to emerge. To achieve this goal, SSOs targeting most of the 79 DMD exons need to be designed. For future development of exon skipping therapy for DMD, there are two key elements: (1) the proper design of SSOs and (2) the application to minor mutation patterns.
First, to design SSOs, it is necessary to confirm exon skipping efficiency in vitro by screening a target region so as to optimize the length and chemistries of SSOs [8, 9]. In previous studies, patient-derived myogenic cells have often been used because they are the only assayable cells to evaluate the restoration of dystrophin protein. In the present study, we assessed the restoration of dystrophin protein with a previously reported SSO (SSO_e50) in the DMD model cells. The result was in agreement with previous studies using patient-derived myogenic cells [12, 13]. We also revealed that DMD exon 50 skipping therapy is applicable to this minor mutation pattern. Therefore, the DMD model cells can serve as an alternative system to evaluate exon skipping of SSOs at the protein level. In addition, the DMD model cells originated from an immortalized cell line and are therefore more suitable than primary skeletal muscle cells for screening large numbers of SSOs. Primary cells have been indicated to have problems with lot-to-lot variations. Recent reports have pointed out that primary skeletal muscle cells need to be immortalized for better detection of proteins [14, 18].
Second, there are significant hurdles to obtain patient cells, because DMD patients have unique mutation patterns in the DMD gene [4]. Besides, the current diagnosis system does not recommend muscle biopsy when MLPA analysis can reveal the deletion or duplication mutations [19]. In the present study, we established a DMD model cell line that has one of the minor mutation patterns: exon 51–57 deletion mutation in the DMD gene. We then revealed that DMD exon 50 skipping therapy is applicable for this minor mutation pattern. According to a previous report, nearly 70% of DMD patients have large deletions in the DMD gene, and among of them, 80% of deletions cover the exon 45–55 (66%) or exon 2–20 (14%) regions [4]. Therefore, many previous studies have focused on therapy for DMD patients who have mutations in hotspot regions, such as with eteplirsen, the SSO targeting DMD exon 51, recently approved by the FDA [32]. The other antisense-based drugs that have been registered in clinical trials are also designed for mutations in hotspot regions, such as DMD exon 44, 45, and 53 (https://clinicaltrials.gov/) [8]. On the other hand, patients with a deletion mutation outside of the hotspot regions (minor groups) may not be treatable with available targeted SSOs. To broaden the application of exon skipping therapy for DMD patients, it is necessary to perform further screening tests of SSOs for those with minor mutations. CRISPR/Cas9-mediated gene editing of the DMD gene in the RD cell line should enable us to establish other assay cell lines that have various deletion patterns, including minor mutations, and to utilize them for the development of exon skipping therapy.
Although this study is only the first attempt to construct an assay system for the evaluation of DMD exon skipping in the RD cell line, in theory, this methodology can be applied to other genetic deletions in the DMD gene. Overall, these DMD model cells can greatly aid in the confirmation of exon skipping efficiency of SSOs targeting the DMD gene before performing in vivo examinations or clinical trials.
References
Pandey SN, Kesari A, Yokota T, Pandey GS. Muscular dystrophy: disease mechanisms and therapies. Biomed Res Int. 2015;2015:456348.
Koenig M, Monaco AP, Kunkel LM. The complete sequence of dystrophin predicts a rod-shaped cytoskeletal protein. Cell. 1988;53:219–28.
Zubrzycka-Gaarn EE, Bulman DE, Karpati G, Burghes AH, Belfall B, Klamut HJ, et al. The Duchenne muscular dystrophy gene product is localized in sarcolemma of human skeletal muscle. Nature. 1988;333:466–9.
Bladen CL, Salgado D, Monges S, Foncuberta ME, Kekou K, Kosma K, et al. The TREAT-NMD DMD global database: analysis of more than 7000 Duchenne muscular dystrophy mutations. Hum Mutat. 2015;36:395–402.
Lee JJA, Yokota T. Translational research in nucleic acid therapies for muscular dystrophies. In: translational research in muscular dystrophy. Tokyo: Springer; 2016. p. 87–102.
Kole R, Williams T, Cohen L. RNA modulation, repair and remodeling by splice switching oligonucleotides. Acta Biochim Pol. 2004;51:373–8.
Dominski Z, Kole R. Restoration of correct splicing in thalassemic pre-mRNA by antisense oligonucleotides. Proc Natl Acad Sci USA. 1993;90:8673–7.
Shimizu-Motohashi Y, Miyatake S, Komaki H, Takeda S, Aoki Y. Recent advances in innovative therapeutic approaches for Duchenne muscular dystrophy: from discovery to clinical trials. Am J Transl Res. 2016;8:2471–89.
Shimo T, Tachibana K, Saito K, Yoshida T, Tomita E, Waki R, et al. Design and evaluation of locked nucleic acid-based splice-switching oligonucleotides in vitro. Nucleic Acids Res. 2014;42:8174–87.
Aartsma-Rus A, van Vliet L, Hirschi M, Janson AAM, Heemskerk H, de Winter CL, et al. Guidelines for antisense oligonucleotide design and insight into splice-modulating mechanisms. Mol Ther. 2009;17:548–53.
Disterer P, Kryczka A, Liu Y, Badi YE, Wong JJ, Owen JS, et al. Development of therapeutic splice-switching oligonucleotides. Hum Gene Ther. 2014;25:587–98.
Aartsma-Rus A, Bremmer-Bout M, Janson AAM, den Dunnen JT, van Ommen G-JB, van Deutekom JCT. Targeted exon skipping as a potential gene correction therapy for Duchenne muscular dystrophy. Neuromuscul Disord. 2002;12:S71–7.
Aartsma-Rus A, Janson AA, Kaman WE, Bremmer-Bout M, den Dunnen JT, Baas F, et al. Therapeutic antisense-induced exon skipping in cultured muscle cells from six different DMD patients. Hum Mol Genet. 2003;12:907–14.
Goyenvalle A, Babbs A, van Ommen G-JB, Garcia L, Davies KE. Enhanced exon-skipping induced by U7 snRNA carrying a splicing silencer sequence: promising tool for DMD therapy. Mol Ther. 2009;17:1234–40.
Echigoya Y, Lim KRQ, Trieu N, Bao B, Miskew B, Vila MC et al. Quantitative antisense screening and optimization for exon 51 skipping in Duchenne muscular dystrophy. Mol Ther. 2017;25:2561–72.
Nguyen Q, Yokota T. Immortalized muscle cell model to test the exon skipping efficacy for Duchenne muscular dystrophy. J Pers Med. 2017;7:E13.
Aartsma-Rus A, Van Deutekom JC, Fokkema IF, Van Ommen GJ, Den Dunnen JT. Entries in the Leiden Duchenne muscular dystrophy mutation database: an overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle Nerve. 2006;34:135–44.
Echigoya Y, Mouly V, Garcia L, Yokota T, Duddy W. In silico screening based on predictive algorithms as a design tool for exon skipping oligonucleotides in Duchenne muscular dystrophy. PLoS ONE. 2015;10:e0120058.
Bushby K, Finkel R, Birnkrant DJ, Case LE, Clemens PR, Cripe L, et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 2010;9:77–93.
Sazani P, Kole R. Sarepta therapeutics, assignee multiple exon skipping compositions for DMD. US patent US9234198B1, 2016 Jan 12.
Aguanno S, Bouche M, Adamo S, Molinaro M. 12-O-tetradecanoylphorbol-13-acetate-induced differentiation of a human rhabdomyosarcoma cell line. Cancer Res. 1990;50:3377–82.
Bouche M, Senni MI, Grossi AM, Zappelli F, Polimeni M, Arnold HH, et al. TPA-induced differentiation of human rhabdomyosarcoma cells: expression of the myogenic regulatory factors. Exp Cell Res. 1993;208:209–17.
Sakuma T, Woltjen K. Nuclease-mediated genome editing: At the front-line of functional genomics technology. Dev Growth Differ. 2014;56:2–13.
Li ZY, Yang J, Gao X, Lu JY, Zhang Y, Wang K, et al. Sequential recruitment of PCAF and BRG1 contributes to myogenin activation in 12-O-tetradecanoylphorbol-13-acetate-induced early differentiation of rhabdomyosarcoma-derived cells. J Biol Chem. 2007;282:18872–8.
Ye J, Coulouris G, Zaretskaya I, Cutcutache I, Rozen S, Madden TL. Primer-BLAST: a tool to design target-specific primers for polymerase chain reaction. BMC Bioinformatics. 2012;13:134.
McAllister RM, Melnyk J, Finkelstein JZ, Adams EC Jr., Gardner MB. Cultivation in vitro of cells derived from a human rhabdomyosarcoma. Cancer. 1969;24:520–6.
Maruyama R, Echigoya Y, Caluseriu O, Aoki Y, Takeda S, Yokota T. Systemic delivery of morpholinos to skip multiple exons in a dog model of Duchenne muscular dystrophy. Methods Mol Biol. 2017;1565:201–13.
Ousterout DG, Kabadi AM, Thakore PI, Majoros WH, Reddy TE, Gersbach CA. Multiplex CRISPR/Cas9-based genome editing for correction of dystrophin mutations that cause Duchenne muscular dystrophy. Nat Commun. 2015;6:6244.
Young CS, Hicks MR, Ermolova NV, Nakano H, Jan M, Younesi S, et al. A single CRISPR-Cas9 deletion strategy that targets the majority of DMD patients restores dystrophin function in hiPSC-derived muscle cells. Cell Stem Cell. 2016;18:533–40.
Nelson CE, Hakim CH, Ousterout DG, Thakore PI, Moreb EA, Castellanos Rivera RM, et al. In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science. 2016;351:403–7.
Tabebordbar M, Zhu K, Cheng JK, Chew WL, Widrick JJ, Yan WX, et al. In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science. 2016;351:407–11.
Stein CA, Castanotto D. FDA-approved oligonucleotide therapies in 2017. Mol Ther. 2017;25:1069–75.
Acknowledgements
TS was supported by Grant-in-Aid for JSPS Research Fellow Grant Number 15J05689. SO was supported by the Basic Science and Platform Technology Program for Innovative Biological Medicine from Japan Agency for Medical Research and Development (AMED). TY was supported by the Friends of Garrett Cumming Research Chair Fund, the HM Toupin Neurological Science Research Chair Fund, Muscular Dystrophy Canada, the Canada Foundation for Innovation (CFI), Alberta Advanced Education and Technology (AET), Canadian Institutes of Health Research (CIHR), Jesse’s Journey – The Foundation for Gene and Cell Therapy, the University of Alberta Faculty of Medicine and Dentistry, and the Women and Children’s Health Research Institute (WCHRI).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Shimo, T., Hosoki, K., Nakatsuji, Y. et al. A novel human muscle cell model of Duchenne muscular dystrophy created by CRISPR/Cas9 and evaluation of antisense-mediated exon skipping. J Hum Genet 63, 365–375 (2018). https://doi.org/10.1038/s10038-017-0400-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s10038-017-0400-0
This article is cited by
-
Production of Duchenne muscular dystrophy cellular model using CRISPR-Cas9 exon deletion strategy
Molecular and Cellular Biochemistry (2023)
-
Applications of CRISPR/Cas9 in the research of malignant musculoskeletal tumors
BMC Musculoskeletal Disorders (2021)
-
Genome-editing approaches and applications: a brief review on CRISPR technology and its role in cancer
3 Biotech (2021)
-
Synthetic cannabinoid CP-55,940 induces apoptosis in a human skeletal muscle model via regulation of CB1 receptors and l-type Ca2+ channels
Archives of Toxicology (2021)
