
Abstract
Cerebrotendinous xanthomatosis (CTX) is likely to be underdiagnosed and precise epidemiological characteristics of CTX are largely unknown as knowledge on the disorder is based mainly on case reports. We conducted a nationwide survey on CTX to elucidate the frequency, clinical picture, and molecular biological background of Japanese CTX patients. In this first Japanese nationwide survey on CTX, 2541 questionnaires were sent to clinical departments across Japan. A total of 1032 (40.6%) responses were returned completed for further analysis. Forty patients with CTX (50.0% male) were identified between September 2012 and August 2015. The mean age of onset was 24.5 ± 13.6 years, mean age at diagnosis was 41.0 ± 11.6 years, and corresponding mean duration of illness from onset to diagnosis was 16.5 ± 13.5 years. The most common initial symptom was tendon xanthoma, followed next by spastic paraplegia, cognitive dysfunction, cataract, ataxia, and epilepsy. The most predominant mutations in the CYP27A1 gene were c.1214G> A (p.R405Q, 31.6%), c.1421G> A (p.R474Q, 26.3%), and c.435G> T (p.G145=, 15.8%). Therapeutic interventions that included chenodeoxycholic acid, HMG-CoA reductase inhibitor, and LDL apheresis reduced serum cholestanol level in all patients and improved clinical symptoms in 40.5% of patients. Although CTX is a treatable neurodegenerative disorder, our nationwide survey revealed an average 16.5-year diagnostic delay. CTX may be underdiagnosed in Japan, especially during childhood. Early diagnosis and treatment are essential to improve the prognosis of CTX.
Similar content being viewed by others


Introduction
Cerebrotendinous xanthomatosis (CTX, MIM# 213700) is an autosomal recessive lipid storage disease caused by mutation of the CYP27A1 gene encoding the mitochondrial cytochrome P 450 enzyme, sterol 27-hydroxylase (CYP27A1, EC 1.14.15.15), which plays a key role in the conversion of cholesterol to bile acids [1, 2]. To date, over 80 pathogenic variants of the CYP27A1 gene have been reported worldwide [3,4,5,6,7,8,9]. Deficiency of CYP27A1 results in impaired bile acid synthesis and increased production of cholesterol metabolites such as cholestanol that accumulates mainly in the brain, tendons, lenses, and vessels [1]. CTX is characterized by neonatal cholestatic jaundice, chronic unexplained diarrhea, juvenile cataract, juvenile osteoporosis, tendon xanthoma, coronary artery disease, and such neurological symptoms as mental retardation/cognitive impairment, spastic paraplegia, ataxia, parkinsonism, peripheral neuropathy, and epilepsy [3, 9,10,11,12,13]. The broad and diverse nature of clinical symptoms in CTX reportedly cause a substantial diagnostic delay of 20–25 years [3, 12, 13]. Moreover, CTX is likely to be underdiagnosed and precise epidemiological characteristics of CTX are largely unknown as knowledge on the disorder is based mainly on case reports. Although several large series of CTX patients have been reported from the Netherlands [3], Italy [13], and the United States [9, 11], only one nationwide survey on this condition has been performed, in which 25 Spanish CTX patients were described [12]. Understanding the current status of CTX and better characterization of the disease are of great importance as CTX is considered a treatable metabolic disorder. We herein report the results of the first nationwide survey on Japanese CTX that identified 40 patients.
Materials and methods
Nationwide survey
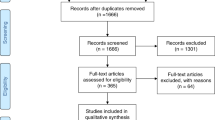
A national survey on CTX was conducted to elucidate the frequency, clinical picture, and molecular biological background of Japanese CTX patients. The target population was patients with CTX who had visited affiliate hospitals between September 2012 and August 2015. A primary questionnaire was sent to 2541 neurology, cardiology, and pediatrics departments across Japan, and 1032 (40.6%) returned completed (Fig. 1). The first survey inquired only on the number of patients with suspected or confirmed CTX who had visited the hospital over the three-year study period. If such a case was identified, a second questionnaire on the detailed clinical features of each patient was sent (Fig. 1).

Flowchart for the present study
The second questionnaire included the parameters of gender, ethnic background, family history, initial symptoms, age at onset, age at diagnosis, age at the latest examination, presenting symptoms and onset age of each symptom, modified Rankin Scale score at the latest examination, CYP27A1 genotype, serum cholestanol and cholesterol levels, results for brain and spinal magnetic resonance imaging (MRI), electroencephalography (EEG), nerve conduction study (NCV), carotid artery ultrasonography, coronary angiography, coronary computed tomography angiography (CTA), ankle-brachial index (ABI), and bone mineral density (BMD), type of treatment, adverse events during treatment, and treatment response. If a particular examination was performed more than once, the latest result was adopted, except for serum cholestanol, for which the serum levels before and after treatment were employed. This study was approved by the Ethical Committee of Shinshu University School of Medicine (approval numbers: 3221 and 3316).
Diagnosis of CTX and definition of clinical phenotypes
A diagnosis of CTX was made if the patient fulfilled the criteria of definite or probable CTX described in Table 1. Since there were no generally accepted diagnostic criteria for CTX, we established original criteria in this study that included clinical symptoms, biochemical findings, genetic testing, and differential diagnosis (Table 1). CTX was categorized into three phenotypes: classical form, spinal form, and non-neurological form. Classical form CTX patients exhibited neurological symptoms attributed to the brain (cerebrum, cerebellum, and/or brainstem). Spinal form CTX patients displayed neurological symptoms limited to spinal cord (i.e., chronic myelopathy), while non-neurological form CTX patients were without any apparent neurological symptoms. In the non-neurological form CTX, patients without genetic testing were excluded from the study, as differential diagnosis from sitosterolemia was difficult without genetic testing.
Statistical analysis
Statistical comparisons between the early-onset (age at onset: <15 years) and late-onset (age at onset: ≥15 years) groups were performed using Fisher’s exact test for binary outcomes and the Wilcoxon rank sum test for continuous variables. Statistical comparisons among the clinical phenotypes (classical, spinal, and non-neurological form) were performed using Fisher’s exact test for binary outcomes and the Steel–Dwass test for continuous variables. Statistical comparison of serum cholestanol levels before and after treatment were carried out using the paired t-test. In all analyses, P < 0.05 was taken to indicate statistical significance.
Results
Demographic features of Japanese CTX patients
A total of 49 cases of suspected or confirmed CTX patients were reported from all over Japan (Fig. 1). Among them, 40 (20 male and 20 female) patients fulfilled the criteria of definite (18 subjects) or probable (22 subjects) CTX (Table 1) and were analyzed for detailed clinical information (Fig. 1). Most patients (n = 34, 85.0%) were reported from a neurology department, with the remaining patients being identified by departments of metabolism/endocrinology (n = 3), cardiology (n = 1), orthopedics (n = 1), or dermatology (n = 1). No patients were reported from a pediatrics department. Although we sent questionnaires to neurology, cardiology, and pediatrics departments, five patients were reported from other departments, (i.e., metabolism/endocrinology, orthopedics, and dermatology). As these patients were managed by several departments, the questionnaires were completed and returned by the departments mainly in charge of the patients.
Seven patients (17.5%) had a family history of parental consanguinity and 10 (25.0%) possessed a family history suggestive of CTX. The age of onset ranged from 0 to 50 years (mean ± standard deviation [SD]: 24.5 ± 13.6 years, Fig. 2a) and the age at diagnosis ranged from 5 to 71 years (mean ± SD: 41.0 ± 11.6 years, Fig. 2b). Although 11 patients developed the disease before 15 years of age, only one was diagnosed as having CTX before that time point (Fig. 2a, b). The duration of illness from onset to diagnosis ranged from 0 to 57 years (mean ± SD: 16.5 ± 13.5 years) and displayed a bimodal distribution (Fig. 2c). The duration of illness from onset to diagnosis in patients who developed CTX at <15 years old (mean ± SD: 27.1 ± 16.8 years) was significantly longer than that in patients who manifested the disease at ≥15 years old (mean ± SD: 12.4 ± 9.6 years, P = 0.008). Patient age at the latest hospital visit ranged from 16 to 79 years (mean ± SD: 47.0 ± 13.9 years). The mean ± SD modified Rankin Scale score was 2.8 ± 1.5 in the cohort. No deaths were reported over the 3-year study period.

Distribution of age at onset a, age at diagnosis b, and duration of illness from onset to diagnosis c among Japanese cerebrotendinous xanthomatosis patients
Clinical features
The most common initial symptom was tendon xanthoma (n = 17, 42.5%, Supplemental Fig. 1A), followed next by spastic paraplegia (n = 13, 32.5%), mental retardation/cognitive impairment (n = 12, 30.0%), juvenile cataract (n = 10, 25.0%), cerebellar ataxia (n = 6, 15.0%), epilepsy (n = 3, 7.5%), parkinsonism/dystonia (n = 3, 7.5%), sensory disturbance attributed to spinal cord/disturbance of posterior column (n = 2, 5.0%), peripheral neuropathy (n = 1, 2.5%), and juvenile osteoporosis/pathologic fracture (n = 1, 2.5%). At the time of survey, the main clinical findings present in descending order of frequency were tendon xanthoma (n = 38, 95.0%), mental retardation/cognitive impairment (n = 28, 70.0%), spastic paraplesia (n = 26, 65.0%), juvenile cataract (n = 24, 60.0%), cerebellar ataxia (n = 17, 42.5%), sensory disturbance attributed to spinal cord/disturbance of posterior column (n = 13, 32.5%), coronary artery disease (n = 8, 20.0%), chronic unexplained diarrhea (n = 8, 20.0%), parkinsonism/dystonia (n = 8, 20.0%), peripheral neuropathy (n = 5, 12.5%), epilepsy (n = 4, 10.0%), and juvenile osteoporosis/pathologic fracture (n = 4, 10.0%). Other findings included stroke, neurogenic bladder, cardiomyopathy, and chronic renal failure in one patient (2.5%) each (Fig. 3). Arteriosclerosis obliterans was not observed in any patients. The most common site of tendon xanthoma was the Achilles tendon (n = 27, 67.5%), followed next by the patellar tendon (n = 7, 17.5%), elbow (n = 3, 7.5%), and heel (n = 2, 5.0%). While many CTX patients displayed mental retardation, epilepsy, and diarrhea at a young age, the occurrence of coronary artery disease and osteoporosis/pathologic fracture was more frequently observed in middle age (Fig. 3).

Frequency of symptoms and onset of each symptom among Japanese cerebrotendinous xanthomatosis patients. N/A data on age of onset was not available
Laboratory, radiological, and physiological findings
All CTX patients exhibited increased serum cholestanol level (mean ± SD: 21.1 ± 10.5 μg/mL, range: 5.8–49.6 μg/mL, normal: 2.4 ± 0.7 μg/mL), which was compatible with previous CTX series of different ethnic background [3, 9, 11,12,13].
All subjects were subjected to brain MRI. Abnormal hyperintense signals on T2-weighted and/or fluid-attenuated inversion recovery (FLAIR) images were detected in 34 (85.0%) patients Supplemental Fig. 1B, C). Primary sites of abnormal intensity were the dentate nucleus of the cerebellum (n = 25, 62.5%), cerebral white matter (n = 17, 42.5%), corticospinal tract (n = 13, 32.5%), cerebellar white matter (n = 8, 20.0%), substantia nigra (n = 4, 10.0%), corpus striatum (n = 2, 5.0%), globus pallidus (n = 2, 5.0%), and thalamus (n = 2, 5.0%). Atrophy of the cerebrum, cerebellum, and brainstem was observed in 40.0% (n = 16), 45.0% (n = 18), and 22.5% (n = 9) of patients, respectively. MRI angiography was evaluated in 31 patients but displayed no abnormal findings, such as cerebrovascular stenosis/occlusion. Carotid artery ultrasonography of 14 patients disclosed increased intima-media thickness in three but no stenosis or occlusion of the carotid artery. Spinal cord MRI of 21 patients revealed abnormal hyperintense signals on T2-weighted images in eight patients. Abnormal intensity was located mainly in the cervical cord (n = 8), followed next by the thoracic cord (n = 4) and lumbar cord (n = 2), at the dorsal and/or lateral column. One patient exhibited an abnormal signal at the lumbar nerve roots. EEG was performed on 25 patients and revealed wave slowing in 13 patients and epileptic discharge in two. NCV testing of 26 patients showed abnormal findings in 12 as decreased motor conduction velocity (n = 7), prolonged distal motor latency (n = 6), decreased amplitude of compound muscle action potential (n = 5), decreased sensory conduction velocity (n = 5), and decreased amplitude of sensory nerve action potential (n = 4).
The coronary artery was evaluated in eight patients (coronary angiography: six patients, coronary CTA: two patients) to disclose stenosis (≥50%) in five. The BMD of 14 patients relative to age-matched normal values was 75.8 ± 17.6% in the lumbar spine and 84.7 ± 9.8% in the femoral neck. ABI assessed in 11 patients was normal in all cases.
CYP27A1 genotype
Genetic analysis of the CYP27A1 gene performed on 19 patients revealed 10 different variants. The most common variant in Japanese CTX patients was c.1214G> A (p.R405Q, 31.6%), followed by c.1421G> A (p.R474Q, 26.3%), c.435G> T (p.G145=, 13.2%), c.1420C> T (p.R474W, 7.9%), c.1176_1117delGA (5.3%), c.254A> G (p.Q85R, 2.6%), c.673C> T (p.R225C, 2.6%), c.1004C> T (p.A335V, 2.6%), c.1238T> A (p.V413D, 2.6%), and c.1342_1343insCACC (2.6%). Two frameshift variants, c.1176_1117delGA and c.1342_1343insCACC, have not been described previously. Although c.673C> T was registered as a benign variant at the National Center for Biotechnology Information (https://www.ncbi.nlm.nih.gov/clinvar/variation/256798/), one patient with compound heterozygosity of c.673C> T (p.R225C) and c.1421G> A (p.R474Q) showed markedly elevated serum cholestanol level (30.7 μg/ml) and relevant clinical findings, including tendon xanthoma and coronary artery disease. The potential impact of the c.673C> T variant on the structure and function of CYP27A1 was assessed using the Polymorphism Phenotyping-2 (PolyPhen-2, http://genetics.bwh.harvard.edu/pph2/) bioinformatics tool [14] and determined to be likely damaging (score: 1.00). Therefore, we judged c.673C> T to be a pathogenic variant in CTX.
Clinical phenotype of CTX
The demographic and clinical characteristics of the patient population according to clinical CTX phenotype are summarized in Table 2. There were 30 patients with the classical form, six with the spinal form, and four with the non-neurological form. Spinal form patients tended to develop the disease later in life (mean ± SD age: 32.8 ± 5.5 years) as compared with those exhibiting the classical form (mean ± SD age: 22.5 ± 14.5 years) or the non-neurological form (mean ± SD age: 27.3 ± 11.6 years), although the differences were not statistically significant. With respect to serum cholestanol level, classical form patients showed significantly higher levels (mean ± SD: 24.0 ± 9.5 μg/mL) than did spinal form patients (11.9 ± 5.6 μg/mL, P = 0.03). Moreover, subjects with the classical form of CTX were more likely to have general symptoms, including diarrhea, cataract, and osteoporosis, as compared with the other clinical phenotypes (Table 2). As expected, subjects with the non-neurological form showed significantly lower modified Rankin Scale, as compared with the other clinical phenotypes (vs. classical form: P = 0.003, vs. spinal form: P = 0.03, Table 2). Regarding genotype–phenotype correlations, it appeared that c.1421G> A (p.R474Q), c.1214G> A (p.R405Q), and c.435G> T (p.G145=) had associations with classical, spinal, and non-neurological CTX forms, respectively (Table 2, Fig. 4).

Genotype–phenotype correlation among Japanese cerebrotendinous xanthomatosis patients. The percentage of alleles (number of alleles) associated with each clinical phenotype is shown. a p.R474Q (c.1421G>A). b p.R405Q (c.1214G>A). c p.G145 = (c.435G>T)
Treatment response
Fifteen patients received chenodeoxycholic acid (CDCA) monotherapy, two received 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMGR) inhibitor monotherapy, 18 received combination therapy of CDCA and HMGR inhibitor, and three underwent low-density lipoprotein (LDL) apheresis. LDL apheresis was performed in combination with CDCA alone or with HMGR inhibitor: all three patients received CDCA and two received additional HMGR inhibitor. CDCA dosage ranged from 125 to 900 mg (mean ± SD: 440.6 ± 148.6 mg). All treatment modalities reduced serum cholestanol level, among which CDCA monotherapy, combination CDCA and HMGR inhibitor, and combination LDL apheresis and CDCA/HMGR inhibitor decreased cholestanol significantly (Table 3). Symptomatic improvement (i.e., improvements in gait disturbance, cognitive dysfunction, tremor, spasticity, ataxia, weakness, psychiatric symptoms, and tendon xanthoma) was observed in six patients treated with CDCA, six with combination CDCA and HMGR inhibitor, and two with combination LDL apheresis and CDCA/HMGR inhibitor (Table 3). Four patients discontinued CDCA due to liver injury.
Discussion
The present study describes the results of the first nationwide survey on CTX in Japan and identifies 40 CTX patients over a three-year study period from September 2012 to August 2015. Although CTX has been considered a rare hereditary disease, this disorder is likely underdiagnosed according to a recent epidemiological study based on the ExAC cohort, a large cohort of over 60,000 unrelated adults, that estimates the incidence of CTX to be 1:134,970–1:461,358 in Europeans, 1:263,222–1:468,624 in Africans, 1:71,677–1:148,914 in Americans, 1:64,247–1:64,712 in East Asians, and 1:36,072–1:75,601 in South Asians [15]. According to this estimation, the number of Japanese CTX patients is calculated to be between 1961 and 1975, which is approximately 50 times higher than the number of patients identified in our nationwide survey. In addition, there was a considerable diagnostic delay (mean ± SD: 16.5 ± 13.5 years) for CTX in the present survey, which was similar to previous studies [3, 12, 13]. It is therefore probable that CTX is underdiagnosed in Japan, especially during in childhood; in fact, only one patient was diagnosed as having CTX before 15 years of age despite 11 developing the disease before that time point. To our surprise, no CTX patients were reported from among 525 major pediatrics departments surveyed nationwide. The diagnostic delay in patients who developed CTX at <15 years old (mean ± SD: 27.1 ± 16.8 years) was >2 times longer than in patients who manifested symptoms at ≥15 years old (mean ± SD: 12.4 ± 9.6 years). Intriguingly, the duration of illness from onset to diagnosis showed a bimodal distribution (Fig. 2c), suggesting that it may take several decades for a diagnosis if not identified correctly in childhood.
The early diagnosis of CTX is crucial since several disease-modifying therapies, such as CDCA, can reverse metabolic derangement and may prevent or even improve the neurologic dysfunction associated with this disease [11, 16,17,18]. Most importantly, treatment effects may diminish once neurological symptoms are fully established, presumably due to the irreversibility of lesions [19, 20], and so therapy is considered more efficacious if started early in the disease course [12, 21]. Several reports have indicated that prolonged unexplained neonatal cholestasis could be the earliest symptom of CTX [10, 13, 21]. Along with mental retardation, epilepsy, and chronic diarrhea, prolonged unexplained neonatal cholestasis represents a key clinical symptom in diagnosing childhood CTX. However, no CTX patient had a history of prolonged neonatal cholestasis in the present survey. One possible reason for this discrepancy is an ethnic difference among study populations [10, 13, 21]. Alternatively, such a medical history could have been missed since the mean ± SD age of the patients was 47.0 ± 13.9 years at the time of survey and no detailed medical history during the neonatal period was available.
The present survey categorized CTX patients into three clinical phenotypes (classical form, spinal form, and non-neurological form) and sought to elucidate the clinical and molecular characteristics of each variant. The possibility of patients with the non-neurological form later developing neurological symptoms was considered, but two 50-year-old genetically confirmed CTX patients showed no evidence of neurological symptoms even ≥20 years after disease onset. Therefore, we regarded the non-neurological form as a distinct clinical phenotype of CTX. As expected, patients with non-neurological CTX showed modest increases in serum cholestanol and preserved activities of daily living (Table 2). The concept of spinal form CTX, also called spinal xanthomatosis, was proposed by Verrips et al. in 1999 [22]. A total of 25 patients with spinal form CTX have been reported to date [12, 22,23,24,25,26,27,28,29,30,31]. Verrips described this variant as a “slowly progressive, mainly spinal cord syndrome that remained for many years the sole expression of CTX”. Most reported spinal form cases have also displayed various neurological symptoms apart from myelopathy, including seizure, dysarthria, cognitive dysfunction, cerebellar ataxia, and polyneuropathy [12, 22], but four spinal form patients without other neurological symptoms have been reported as well [23,24,25,26]. In the present study, we defined spinal form as CTX patients whose neurological symptoms were limited to the spinal cord and identified six cases, two of which were previously described [23, 24]. Patients in this subgroup exhibited later disease onset and lower frequencies of tendon xanthoma, diarrhea, cataract, and osteoporosis as compared with the classical form (Table 2). Spinal form patients also showed significantly lower serum cholestanol levels as compared with the classical form (Table 2), in contrast to a previous report [12]. A possible reason for this discrepancy may lie in the definition of spinal form CTX: spinal form patients in the present study possessed no neurological symptoms other than myelopathy, while those described by Pilo-de-la-Fuente showed various neurological symptoms other than myelopathy [12].
With regard to CYP27A1 genotype, the present survey revealed that c.1214G> A (p.R405Q), c.1421G> A (p.R474Q), and c.435G> T (p.G145=) were the three most common variants in Japanese CTX, accounting for >70% of variant alleles. Whereas earlier reports indicated no correlation between any given variant and any specific symptom, onset age, or severity [3, 12], this survey revealed possible associations between p.R474Q and classical form CTX, p.R405Q and spinal form CTX, and p.G145= and non-neurological form CTX (Table 2, Fig. 4), although there was considerable phenotypic variation among patients with the same genotype. Greater numbers of genetically confirmed patients will be necessary to elucidate the precise genotype-phenotype correlation of CTX.
Lastly, most patients in the present survey received disease-modifying therapies for CTX, including CDCA, HMGR inhibitor, and/or LDL apheresis. Among them, oral CDCA was the most common treatment. CDCA is believed to inhibit excessive cholestanol synthesis [11, 12, 16, 17, 32], although no statistical analyses of its efficacy have been released to date. We observed that CDCA significantly reduced serum cholestanol level (P = 0.0005, Table 3) and ameliorated neurological symptoms in 40% of patients with CDCA treatment (Table 3). HMGR inhibitor monotherapy reduced serum cholestanol level by 38.3% (Table 3). Combination therapy with CDCA and HMGR inhibitor [33] also significantly reduced serum cholestanol level (P = 0.009, Table 3). However, the percent decrease of cholestanol by combination therapy (59.3%) was not superior to that by CDCA monotherapy (77.6%), and so the efficacy of HMGR inhibitor add-on therapy to CDCA remains uncertain. LDL apheresis in combination with CDCA alone or with HMGR inhibitor was performed on three patients. Serum cholestanol was reduced by approximately half after each round, but later level returned to pre-apheresis levels within 2 weeks, as previously reported [34, 35]. Taking into consideration its relatively high invasiveness and cost, LDL apheresis may be better suited as an add-on therapy for CTX patients unresponsive to CDCA and/or HMGR inhibitors rather than as a stand-alone treatment for CTX [34,35,36,37].
This first nationwide survey might not cover all CTX patients as we surveyed only neurology, cardiology, and pediatrics departments across Japan and the response rate was only modest (40.6%). This study was also limited by the lack of a strict definition of treatment response and information on treatment duration. In addition, data on NCS were insufficiently detailed to determine the type of neuropathy (i.e., demyelination or axonal damage). These issues need resolution in future follow-up surveys.
References
Cali JJ, Hsieh CL, Francke U, Russell DW. Mutations in the bile acid biosynthetic enzyme sterol 27-hydroxylase underlie cerebrotendinous xanthomatosis. J Biol Chem. 1991;266:7779–83.
Cali JJ, Russell DW. Characterization of human sterol 27-hydroxylase. A mitochondrial cytochrome P-450 that catalyzes multiple oxidation reaction in bile acid biosynthesis. J Biol Chem. 1991;266:7774–8.
Verrips A, Hoefsloot LH, Steenbergen GC, Theelen JP, Wevers RA, Gabreels FJ, et al. Clinical and molecular genetic characteristics of patients with cerebrotendinous xanthomatosis. Brain. 2000;123:908–19.
Gallus GN, Dotti MT, Federico A. Clinical and molecular diagnosis of cerebrotendinous xanthomatosis with a review of the mutations in the CYP27A1 gene. Neurol Sci. 2006;27:143–9.
Gallus GN, Dotti MT, Mignarri A, Rufa A, Da Pozzo P, Cardaioli E, et al. Four novel CYP27A1 mutations in seven Italian patients with CTX. Eur J Neurol. 2010;17:1259–62.
Nozue T, Higashikata T, Inazu A, Kawashiri MA, Nohara A, Kobayashi J, et al. Identification of a novel missense mutation in the sterol 27-hydroxylase gene in two Japanese patients with cerebrotendinous xanthomatosis. Intern Med. 2010;49:1127–31.
Chen W, Kubota S, Kim KS, Cheng J, Kuriyama M, Eggertsen G, et al. Novel homozygous and compound heterozygous mutations of sterol 27-hydroxylase gene (CYP27) cause cerebrotendinous xanthomatosis in three Japanese patients from two unrelated families. J Lipid Res. 1997;38:870–9.
Suh S, Kim HK, Park HD, Ki CS, Kim MY, Jin SM, et al. Three siblings with Cerebrotendinous Xanthomatosis: a novel mutation in the CYP27A1 gene. Eur J Med Genet. 2012;55:71–4.
Lee MH, Hazard S, Carpten JD, Yi S, Cohen J, Gerhardt GT, et al. Fine-mapping, mutation analyses, and structural mapping of cerebrotendinous xanthomatosis in U.S. pedigrees. J Lipid Res. 2001;42:159–69.
Clayton PT, Verrips A, Sistermans E, Mann A, Mieli-Vergani G, Wevers R. Mutations in the sterol 27-hydroxylase gene (CYP27A) cause hepatitis of infancy as well as cerebrotendinous xanthomatosis. J Inherit Metab Dis. 2002;25:501–13.
Berginer VM, Salen G, Shefer S. Long-term treatment of cerebrotendinous xanthomatosis with chenodeoxycholic acid. N Engl J Med. 1984;311:1649–52.
Pilo-De-La-Fuente B, Jimenez-Escrig A, Lorenzo JR, Pardo J, Arias M, Ares-Luque A, et al. Cerebrotendinous xanthomatosis in Spain: clinical, prognostic, and genetic survey. Eur J Neurol. 2011;18:1203–11.
Mignarri A, Gallus GN, Dotti MT, Federico A. A suspicion index for early diagnosis and treatment of cerebrotendinous xanthomatosis. J Inherit Metab Dis. 2014;37:421–9.
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–9.
Appadurai V, Debarber A, Chiang PW, Patel SB, Steiner RD, Tyler C, et al. Apparent underdiagnosis of cerebrotendinous xanthomatosis revealed by analysis of ~60,000 human exomes. Mol Genet Metab. 2015;116:298–304.
Van Heijst AF, Verrips A, Wevers RA, Cruysberg JR, Renier WO, Tolboom JJ. Treatment and follow-up of children with cerebrotendinous xanthomatosis. Eur J Pediatr. 1998;157:313–6.
Mondelli M, Sicurelli F, Scarpini C, Dotti MT, Federico A. Cerebrotendinous xanthomatosis: 11-year treatment with chenodeoxycholic acid in five patients. An electrophysiological study. J Neurol Sci. 2001;190:29–33.
Yoshinaga T, Sekijima Y, Koyama S, Maruyama K, Yoshida T, Kato T, et al. Clinical and radiological findings of a cerebrotendinous xanthomatosis patient with a novel p.A335V mutation in the CYP27A1 gene. Intern Med. 2014;53:2725–9.
Peynet J, Laurent A, De Liege P, Lecoz P, Gambert P, Legrand A, et al. Cerebrotendinous xanthomatosis: treatments with simvastatin, lovastatin, and chenodeoxycholic acid in 3 siblings. Neurology. 1991;41:434–6.
Pilo De La Fuente B, Ruiz I, Lopez De Munain A, Jimenez-Escrig A. Cerebrotendinous xanthomatosis: neuropathological findings. J Neurol. 2008;255:839–42.
Pierre G, Setchell K, Blyth J, Preece MA, Chakrapani A, Mckiernan P. Prospective treatment of cerebrotendinous xanthomatosis with cholic acid therapy. J Inherit Metab Dis. 2008;31(Suppl 2):S241–5.
Verrips A, Nijeholt GJ, Barkhof F, Van Engelen BG, Wesseling P, Luyten JA, et al. Spinal xanthomatosis: a variant of cerebrotendinous xanthomatosis. Brain. 1999;122:1589–95.
Abe R, Sekijima Y, Kinoshita T, Yoshinaga T, Koyama S, Kato T, et al. Spinal form cerebrotendinous xanthomatosis patient with long spinal cord lesion. J Spinal Cord Med. 2016;39:726–9.
Yanagihashi M, Kano O, Terashima T, Kawase Y, Hanashiro S, Sawada M, et al. Late-onset spinal form xanthomatosis without brain lesion: a case report. BMC Neurol. 2016;16:21.
Nicholls Z, Hobson E, Martindale J, Shaw PJ. Diagnosis of spinal xanthomatosis by next-generation sequencing: identifying a rare, treatable mimic of hereditary spastic paraparesis. Pract Neurol. 2015;15:280–3.
Saute JA, Giugliani R, Merkens LS, Chiang JP, Debarber AE, De Souza CF. Look carefully to the heels! A potentially treatable cause of spastic paraplegia. J Inherit Metab Dis. 2015;38:363–4.
Schreiner A, Hopen G, Skrede S. Cerebrotendinous xanthomatosis (cholestanolosis). Investigations on two sisters and their family. Acta Neurol Scand. 1975;51:405–16.
Swartz M, Burman KD, Salen G. Cerebrotendinous xanthomatosis: a cause of cataracts and tendon xanthoma. Am J Med Sci. 1982;283:147–52.
Kuriyama M, Fujiyama J, Yoshidome H, Takenaga S, Matsumuro K, Kasama T, et al. Cerebrotendinous xanthomatosis: clinical and biochemical evaluation of eight patients and review of the literature. J Neurol Sci. 1991;102:225–32.
Restuccia D, Di Lazzaro V, Servidei S, Colosimo C, Tonali P. Somatosensory and motor evoked potentials in the assessment of cerebrotendinous xanthomatosis before and after treatment with chenodeoxycholic acid: a preliminary study. J Neurol Sci. 1992;112:139–46.
Dotti MT, Federico A, Signorini E, Caputo N, Venturi C, Filosomi G, et al. Cerebrotendinous xanthomatosis (van Bogaert-Scherer-Epstein disease): CT and MR findings. Am J Neuroradiol. 1994;15:1721–6.
Koopman BJ, Wolthers BG, Van Der Molen JC, Waterreus RJ. Bile acid therapies applied to patients suffering from cerebrotendinous xanthomatosis. Clin Chim Acta. 1985;152:115–22.
Kuriyama M, Tokimura Y, Fujiyama J, Utatsu Y, Osame M. Treatment of cerebrotendinous xanthomatosis: effects of chenodeoxycholic acid, pravastatin, and combined use. J Neurol Sci. 1994;125:22–8.
Ito S, Kuwabara S, Sakakibara R, Oki T, Arai H, Oda S, et al. Combined treatment with LDL-apheresis, chenodeoxycholic acid and HMG-CoA reductase inhibitor for cerebrotendinous xanthomatosis. J Neurol Sci. 2003;216:179–82.
Mimura Y, Kuriyama M, Tokimura Y, Fujiyama J, Osame M, Takesako K, et al. Treatment of cerebrotendinous xanthomatosis with low-density lipoprotein (LDL)-apheresis. J Neurol Sci. 1993;114:227–30.
Berginer VM, Salen G. LDL-apheresis cannot be recommended for treatment of cerebrotendinous xanthomatosis. J Neurol Sci. 1994;121:229–32.
Dotti MT, Lutjohann D, Von Bergmann K, Federico A. Normalisation of serum cholestanol concentration in a patient with cerebrotendinous xanthomatosis by combined treatment with chenodeoxycholic acid, simvastatin and LDL apheresis. Neurol Sci. 2004;25:185–91.
Acknowledgements
The authors thank Y. Iwanaka, S. Ito, T. Yoshida, K. Tokuoka, H. Yuasa, M. Takata, S. Hirose, S. Togashi. H. Taniguchi, K. Ishihara, T. Hashimoto, T. Takeshima, S. Akazawa, A. Haraguchi, M. Ito, K. Takase, S. Susa, Y. Honma, H. Kaneko, Y. Touma, K. Yamamoto, Y. Harigaya, K. Tamura, I. Yokota, T. Kawazoe, S. Takagi, T. Hamada, M. Takemoto, H. Nagayama, M. Mizuno, K. Uemura, Y. Kajimoto, and O. Kano for providing clinical patient information. This study was supported by a grant from the Research Committee for Cerebrotendinous Xanthomatosis, Research on Measures against Intractable Diseases by the Japanese Ministry of Health, Labour, and Welfare and a grant from the Research Committee for Primary Hyperlipidemia, Research on Measures against Intractable Diseases by the Japanese Ministry of Health, Labour, and Welfare.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Sekijima, Y., Koyama, S., Yoshinaga, T. et al. Nationwide survey on cerebrotendinous xanthomatosis in Japan. J Hum Genet 63, 271–280 (2018). https://doi.org/10.1038/s10038-017-0389-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s10038-017-0389-4
This article is cited by
-
Cerebrotendinous Xanthomatosis patients with late diagnosed in single orthopedic clinic: two novel variants in the CYP27A1 gene
Orphanet Journal of Rare Diseases (2024)
-
Case of cerebrotendinous xanthomatosis with giant xanthomas and literature review
The Egyptian Journal of Neurology, Psychiatry and Neurosurgery (2023)
-
Differing clinical features between Japanese siblings with cerebrotendinous xanthomatosis with a novel compound heterozygous CYP27A1 mutation: a case report
BMC Neurology (2022)
-
Cerebellar Cognitive Affective Syndrome in a Case of Cerebrotendinous Xanthomatosis
The Cerebellum (2022)
-
Clinical and genetic characteristics of Chinese patients with cerebrotendinous xanthomatosis
Orphanet Journal of Rare Diseases (2019)
