
Abstract
Using genome-editing technologies to correct specific mutations represents a potentially transformative new approach for treating genetic disorders. Despite rapid advances in the field of genome editing, it is still unclear whether the long-standing goal of in vivo targeted transgene integration is feasible. This is primarily because current tools are inefficient. In particular, current technologies are incapable of targeted gene knock-in in non-dividing cells, the major building blocks of adult tissues. This poses a significant barrier for developing therapeutic strategies to treat a broad range of devastating genetic disorders. Recently, our group has developed a unique CRISPR/Cas9-based strategy, termed homology-independent targeted insertion (HITI), which enables targeted gene insertion in non-dividing cells, both in vitro and in vivo. This review will summarize current progress in developing this technology, and discuss the potential impact of HITI-based gene-correction therapies.
Similar content being viewed by others

Introduction
The intracellular delivery of wild-type genetic sequences is one of the most promising therapeutic approaches for reverting detrimental phenotypes caused by loss-of-function mutations. Viral-mediated gene replacement therapy is one way of achieving this goal, and is a promising treatment option for many inherited diseases. In fact, clinical trial using this technology have revealed remarkable therapeutic benefits [1]. However, the major limitations of this method are incomplete control over transgene copy numbers and expression levels, as well as risk of adverse events (e.g., insertional mutagenesis or activation of (proto)-oncogenes)[1]. In addition, the duration of therapeutic benefit, which is influenced by levels of transgene expression and activity, is also unpredictable, particularly when non-integrating vectors are used. For example, the initial results of a clinical trial for Leber’s congenital amaurosis, a blindness disease, indicated that non-integrating adeno-associated viral (AAV) vector delivery of transgenes was safe and showed therapeutic efficacy in repairing human visual function [2, 3]. However, recently published 3-year follow-up data from the same and other groups demonstrated that the initial gains in retinal sensitivity waned over time and were not successful in achieving meaningful improvements in objective measures of visual function [4, 5]. These results show that although non-integrating vector-based gene therapy is safe and effective during the initial stage of treatment, the therapeutic effects are not permanent. Furthermore, this gene-complementation strategy cannot be used to treat gain-of-function genetic mutations.
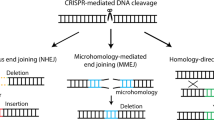
Recent advances in the genome-editing field have revolutionized methods for generating novel genetic resources for biomedical research. It is now possible to exploit the homology-directed repair (HDR) pathway to realize site-specific transgene integration; this represents the most promising way of overcoming limitations of current gene-replacement therapies [6,7,8]. HDR has been extensively used to replace both loss-of-function and gain-of-function alleles with wild-type sequences, thereby recovering gene function and eliminating pathogenic effects. In theory, the therapeutic effect should be permanent once the disease-causing mutation has been fixed. However, the utility of HDR-based approaches is limited by low efficiency in most primary cell types. Moreover, HDR only occurs during the S/G2 phases of the cell cycle, making it inaccessible to non-dividing cells, which are prevalent in post-natal animal tissues [9, 10]. The other major DNA double-strand break (DSB) repair pathway, non-homologous end joining (NHEJ), ligates DNA ends directly and is active throughout the cell cycle in a large variety of adult cells, including both proliferating and post-mitotic cells [11]. In addition, in most higher organisms, NHEJ activity far exceeds HDR activity [11]. These advantages indicate that harnessing the NHEJ pathway for targeted knock-in may represent a viable way of overcoming technical hurdles associated with HDR-based approaches (Fig. 1). Here we review NHEJ-based targeted gene knock-in methods, and in vivo applications.

CRISPR/Cas9-mediated targeted genome editing. Streptococcus pyogenes Cas9 (SpCas9) forms a complex with a single-guide RNA (sgRNA) that recognizes a specific 20-bp homologous sequence in front of the NGG PAM sequence. Once bound, these complexes induce double strand breaks (DSBs) at targeted genomic loci. These DSBs are then repaired by intracellular DSB repair machineries that can be categorized into two major types: (1) error-free homology-directed repair (HDR), which repairs DSBs using a homologous chromatid or chromosome, and (2) error-prone non-homologous end joining (NHEJ), which processes and ligates DNA ends directly. Error-prone NHEJ often introduces indels, which can disrupt the target gene. In contrast, in the presence of ectopic homologous DNA, HDR can introduce any sequence into the target site. HDR only occurs during the S-G2 phase of the cell cycle; whereas NHEJ is active throughout the cell cycle in a variety of adult cell types, making it accessible to non-dividing cells. Homology-independent targeted integration (HITI) hijacks the NHEJ pathway and inserts ectopic DNA at target sites in both dividing and non-dividing cells
NHEJ-mediated targeted gene knock-in method
In 2013, two groups reported the generation of DSBs at a targeted genomic locus and a donor plasmid using zinc finger nuclease (ZFN) and transcription activator-like effector nucleases (TALEN), one type of engineered nucleases that facilitate the introduction of exogenous DNA sequence into the genome of cell lines via homology-independent NHEJ (Table 1) [12, 13]. Similar methods relying on homology-independent NHEJ-based DNA ligation have been proposed for transgene insertion in various organisms [14,15,16,17,18,19,20,21,22,23,24,25,26]. All of these reports employed the approach that digestion of both donor and chromosomal target allows for large DNA insertion (up to 50 kb) into the genomic DSB site (i.e. target site). However, as the NHEJ-mediated knock-in method is homology-independent, the direction in which the ectopic DNA fragment is inserted cannot be controlled. Furthermore, because it is necessary to process DNA ends prior to ligate the break ends when the generated DSBs are non-complementary ends, NHEJ has long been considered to be error-prone, as targeted gene knockout via error-prone NHEJ is highly efficient in many organisms [27]. HDR can replace the target sequence, whereas NHEJ-mediated strategy can add an ectopic DNA sequence at target locus instead of replacing the original genomic sequence. Because of these studies, researchers were concerned about the feasibility of using NHEJ-mediated gene knock-in methods over classical HDR-mediated targeted integration methods.
In 2016, our group devised a fundamentally improved NHEJ-based homology-independent strategy for targeted transgene integration that used the CRISPR/Cas9 system; we named it homology-independent targeted integration (HITI) (Fig. 1) [28]. CRISPR/Cas9 is a recently developed RNA-guided targeted DNA nuclease tool. A complex of Cas9 and guide RNA (gRNA) recognizes and introduces DSBs at specific 20-bp target genome sequences recognized by the gRNA [29,30,31]. We hypothesized that the error-free NHEJ pathway (rather than error-prone NHEJ) is the predominant pathway, thereby resulting in precise and efficient targeted gene knock-in. To prove the feasibility of using NHEJ-mediated gene knock-in methods over classical HDR-based knock-in, we first improved upon existing NHEJ-based methods and developed CRISPR/Cas9-based HITI method. Based on side-by-side comparison in dividing HEK293 cells using CRISPR/Cas9, we demonstrated that the efficiency of NHEJ-mediated HITI is 10 times higher than for HDR. Sequencing result showed that upon integration, the majority of junction sites were free of insertions or deletions (indels), suggesting dominance of error-free repair in this context. In the same experiment, we also used gRNAs to specify the direction of insertion (Fig. 2), resulting in only 2.1% of the cases was the cassette inserted in the opposite direction. This suggests that our NHEJ-based HITI method preferentially generates unidirectional knock-ins. Taken together, we have established HITI, which is a robust and unidirectional targeted knock-in method capable of functioning in dividing cell lines.

Schematic of HITI using SpCas9. The SpCas9/sgRNA complex introduces DSBs 3 base pairs upstream of the PAM sequence in the genomic target sequence, resulting in two blunt ends. The exact same SpCas9/sgRNA target sequence is loaded onto the donor DNA in the reverse direction. Targeted genomic loci, as well as the donor DNA, are cleaved by Cas9/gRNA and the linearized donor DNAs are integrated into target sites via the NHEJ DSB repair pathway. If donor DNA is integrated in the correct orientation, junction sequences are protected from further cleavage by Cas9/gRNA. If donor DNA integrates in the reverse orientation, Cas9/gRNA will excise the integrated donor DNA due to the presence of intact Cas9/gRNA target sites. Blue pentagon, Cas9/gRNA target sequence. Black line within blue pentagon, SpCas9 cleavage site. GOI, gene of interest
For classical HDR-based CRISPR/Cas9 genome-editing strategies, cells are transfected with Cas9, gRNA, and donor DNA that contain homology arms specific for the genomic locus of interest. HDR of the Cas9-mediated DSB results in insertion of the genetic material between the homology arms. In the case of HITI, donor plasmids lack homology arms. Consequently, repair of the Cas9-induced genomic DSB cannot occur through the HDR pathway. The donor DNA is designed to include Cas9 cleavage site(s) that flank the donor sequence. Cas9 therefore cleaves both the genomic target sequence and the donor plasmid, thereby generating blunt ends associated with both target and donor sequences. The linearized donor DNA plasmid can then be used for repair by the NHEJ pathway, allowing for its integration into the genomic DSB site. Once incorporated into the genome, donor DNA that inserted in the desired orientation disrupts the Cas9 target sequence and prevents further Cas9 cutting. If the genomic DSB is rejoined by error-free NHEJ without donor DNA insertion, the Cas9 target sequence will remain intact and a second round of Cas9 cutting will ensue (Fig. 2).
Because NHEJ is active throughout the cell cycle, non-dividing cells, which are in the G0/G1 phase, also harbor NHEJ activity. To take advantage of this, we also tested the efficacy of HITI in non-dividing cells in vitro, and demonstrated that NHEJ-mediated HITI is highly efficient in non-dividing cells (~60% of transfected neurons); this was the first demonstration of targeted gene knock-in in a non-dividing cell type [28].
In vivo genome editing via various tools
Because the majority of cells within the adult mammalian body are terminally differentiated, post-mitotic cells (or quiescent stem cells), the classic HDR method is generally inefficient for in vivo applications. There are only a few exceptions to this rule, such as the dividing cells of the liver [6, 32, 33]. Furthermore, there were no established technologies for the targeted knock-in of genetic material in non-dividing cells. As a proof-of-concept for in vivo application, we first transfected HITI components (Cas9, gRNA, and donor plasmids) by either in utero or in vivo electroporation, and successfully achieved targeted transgene insertion in post-mitotic cells of the brain, kidney, and muscle. Despite high efficiency in vitro, the in vivo gene knock-in efficiency was quite low (~1% of cells), being compromised by low DNA transduction efficiency.
To achieve more efficient and on-target in vivo gene delivery, we switched to AAVs [34]. We loaded Cas9, gRNA, and donor DNA into AAV vectors, and packaged them with AAV serotype 8 or 9, which were previously shown to display high infection capability for many organs with therapeutic safety [35, 36]. We delivered AAVs to the visual cortex of the adult mouse brain via local injection, and observed satisfactory in vivo GFP knock-in efficiency in non-dividing cells near the injection site (3.5% of the cells within 300 µm of the injection site) [28]. Similar to the brain, local delivery of AAVs through intramuscular injection in adult mice also resulted in accurate GFP knock-in in skeletal muscle cells. In addition to in situ injections, we also tested systemic delivery. We infected neonatal mice with GFP knock-in AAVs via intravenous injection. Two weeks post-infection, we observed many GFP knock-in cells (3–10% of cell) throughout the heart, muscle, and liver. The high accuracy and low off-target effects of HITI were also carefully validated by single-cell analysis and next generation sequencing.
Recently, several other genome-editing methods have been reported for in vivo application besides HDR and HITI. Microhomology-mediated end-joining (MMEJ) is another end-joining pathway that is activated when short (5–25 bp) homologous sequences anneal to both strands [37]. In 2014, Nakade et al. have developed a novel MMEJ-mediated targeted gene knock-in method and named Precise Integration into Target Chromosome (PITCh) [38]. To insert an exogenous cassette at target locus, the PITCh vector contains very short (5–25 bp) microhomology sequences flanking the both sides of the exogenous cassette [38, 39]. Similar to HITI, both chromosomal target locus and PITCh donor DNA are digested by CRISPR/Cas9 or TALEN. Then, microhomology sequences of PITCh donor are exposed and integrated at the digested chromosomal target site via MMEJ. Recently, another group has demonstrated that MMEJ-mediated PITCh method is applicable for in vivo genome editing [40]. Using this method, they have succeeded in rescuing the lethal phenotype of fumarylacetoacetate hydrolase-deficient mice, a hereditary tyrosinemia type I mouse model. Furthermore, this group devised the vector structure and named the new method homology-mediated end joining-based strategy [41]. They extended the length of homology arms (~800 bp), thus allowing the exogenous cassette to be integrated at target site through either HDR or NHEJ pathway. Using this donor, they have successfully performed targeted gene knock-in in vivo. These methods represent alternative tools of HITI for in vivo gene knock-in.
In vivo genome correction via HITI
Gene replacement therapy via in vivo genome editing would offer several advantages over classical gene therapy strategies by inserting a functional copy of the defective gene into the endogenous locus. Furthermore, in vivo genome editing is better than ex vivo strategies for many reasons, such as the potential to target a wider range of cell types (including those that are difficult to culture) [42]. However, classical HDR-mediated gene correction methods have not been successfully implemented in vivo to edit non-dividing cells [9]. Thanks to the highly efficient error-prone NHEJ pathway, targeted gene disruption in somatic cells is a step closer to in vivo applications. Feng Zhang’s group reported on targeted gene knockouts in post-mitotic neurons via in vivo Cas9 and gRNA delivery, opening the door for in vivo studies of genetic elements underpinning brain functions [43]. More recently, NHEJ-based targeted gene knockout approach has been utilized to improve muscle function in a mouse model of duchenne muscular dystrophy (DMD), by eliminating a disease-causing mutation through Cas9 and dual gRNA mediated disruption of exon 23 [44,45,46]. Furthermore, Bakondi et al. demonstrated that single variant-specific gRNA can generate NHEJ-mediated allele-specific disruption of autosomal dominant point mutation at Rho gene [47]. By in vivo retinal electroporating the mutation-specific gRNA, they prevented retinal degeneration and improved visual function in blindness rat model. However, NHEJ-based targeted disruption strategy has not been widely used in managing target mutation/disease. Nonetheless, these reports suggest that conducting targeted knock-in using NHEJ-mediated HITI may allow us to overcome technical hurdles that plague existing genome-editing technologies in a wide range of tissues and organs in vivo.
As a proof-of-principal for applying HITI as a gene replacement therapy, we used HITI to treat the Royal College of Surgeons (RCS) rat model of retinitis pigmentosa (RP), an inherited degenerative eye disease that is a common cause of blindness in humans [48]. A homozygous mutation in the Mertk gene, namely a 1.9-kb deletion that includes parts of intron 1 and exon 2, results in defective phagocytic function within the retinal pigment epithelium [49]. This leads to degeneration of both the retinal pigment epithelium and overlaying photoreceptors, and eventually blindness. To restore Mertk gene function in the RP retina, we generated an AAV vector to insert a functional copy of exon 2 in front of the deletion site in intron 1. AAV vectors were delivered into the eyes by subretinal injection at postnatal week 3. One month post-injection, we observed partial rescue of disease phenotypes, namely improved morphology of the photoreceptor outer nuclear layer, and improved visual function (as measured by electroretinography). These results demonstrate that HITI treatment can rescue and preserve retinal visual function. Because HITI can be used to target a wide variety of cell types, it is highly likely that this technology is suitable for treating other genetic diseases that affect other non-dividing tissues, such as DMD, Parkinson’s disease and amyotrophic lateral sclerosis.
Challenges for clinical translation
To translate HITI technology into a clinical application, several major barriers must still be overcome. One major barrier is efficiency. Although the current HITI technology can insert DNA at a chosen target site in many non-dividing tissues, its efficiency is less than 5% in most cases. This level of efficiency can result in therapeutic benefit, as seen in the RP retina, but even in this case phenotypic rescue was partial and not sufficient to completely restore vision. As a result, to use HITI technology to treat diseases in the clinic will require much higher gene-correction efficiencies. To improve HITI efficiency, mechanistic study is important for identifying the major regulators of NHEJ during the execution of HITI. These studies will potentially allow us to establish strategies to facilitate NHEJ–HITI activity in target cells. The screen for small molecules and/or siRNAs/shRNAs that can further enhance NHEJ activity represents a straightforward strategy for improving HITI efficiency. It is also important to develop the most suitable in vivo DNA delivery method for specific type of cell/tissue/organ, fulfilling another important factor for the realization of efficient in vivo genome editing. Another concern related to using HITI for clinical applications is safety. Although we have demonstrated that the off-target effects for HITI are minimal (by deep sequencing of predicted off-target loci), further unbiased deep sequencing analyses are needed to comprehensively evaluate HITI off-target effects. In addition to these two major concerns, it is still unclear how frequent and where ectopic DNA fragments integrate into non-target loci. Recently, the improved version of Cas9 (eSpCas9 and SpCas9-HF1) has been developed and demonstrated low off-target effect [50, 51]. By using these high-fidelity Cas9 genes, the safety of HITI-mediated gene therapy might be further improved. Finally, HITI can only insert ectopic DNA into a target locus, but cannot, for example, remove a mutation from the genome. Thus, the types of genetic defects that HITI technology will be able to treat is limited. If HITI-mediated gene replacement strategies could be further developed, this technology has the potential to evolve into a versatile tool kit for gene therapy.
Conclusions
We have developed a robust and versatile NHEJ-mediated targeted gene modification system, which is named HITI. HITI not only facilitates targeted gene knock-in in cultured cells, but also allows for efficient targeted integration of ectopic DNA in vivo (both for dividing and non-dividing cell types). More importantly, HITI has been successfully used to rescue a loss-of-function mutation in a rat model of RP, via the targeted insertion of a functional exon into the disease-associated Mertk gene. In theory, this new tool holds great promise for editing “any gene” in “any cell/tissue/organ” at “any time” in living organisms, thus constituting a revolutionary breakthrough for basic and translational biomedical studies. For example, because HITI can enable in vivo targeted gene knock-in in adult neurons, this system provides an unprecedented opportunity for advancing neuroscience research, and for developing therapies against debilitating disorders of the brain. To reach this potential, it is necessary to improve the current version of HITI, with the goals of achieving higher efficiency, broadening its versatility, and ensuring safety when implemented in the clinical setting. Once these barriers are overcome, HITI technology may open broad new avenues for developing targeted gene therapies.
References
Naldini L. Gene therapy returns to centre stage. Nature. 2015;526:351–60.
Maguire AM, Simonelli F, Pierce EA, Pugh EN, Mingozzi F, Bennicelli J, et al. Safety and efficacy of gene transfer for Leber’s congenital amaurosis. N Engl J Med. 2008;358:2240–8.
Bainbridge JW, Smith AJ, Barker SS, Robbie S, Henderson R, Balaggan K, et al. Effect of gene therapy on visual function in Leber’s congenital amaurosis. N Engl J Med. 2008;358:2231–9.
Jacobson SG, Cideciyan AV, Roman AJ, Sumaroka A, Schwartz SB, Heon E, Hauswirth WW. Improvement and decline in vision with gene therapy in childhood blindness. N Engl J Med. 2015;372:1920–6.
Bainbridge JWB, Mehat MS, Sundaram V, Robbie SJ, Barker SE, Ripamonti C, et al. Long-term effect of gene therapy on leber’s congenital amaurosis. N Engl J Med. 2015;372:1887–97.
Li H, Haurigot V, Doyon Y, Li T, Wong SY, Bhagwat AS, et al. In vivo genome editing restores haemostasis in a mouse model of haemophilia. Nature. 2011;475:217–21.
Genovese P, Schiroli G, Escobar G, Di Tomaso T, Firrito C, Calabria A, et al. Targeted genome editing in human repopulating haematopoietic stem cells. Nature. 2014;510:235–40.
Lombardo A, Cesana D, Genovese P, Di Stefano B, Provasi E, Colombo DF, et al. Site-specific integration and tailoring of cassette design for sustainable gene transfer. Nat Methods. 2011;8:861–9.
Iyama T, Wilson DM. DNA repair mechanisms in dividing and non-dividing cells. DNA Repair. 2013;12:620–36.
Orthwein A, Noordermeer SM, Wilson MD, Landry S, Enchev RI, Sherker A, et al. A mechanism for the suppression of homologous recombination in G1 cells. Nature. 2015;528:422–6.
Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem. 2010;79:181–211.
Maresca M, Lin VG, Guo N, Yang Y. Obligate ligation-gated recombination (ObLiGaRe): custom-designed nuclease-mediated targeted integration through nonhomologous end joining. Genome Res. 2013;23:539–46.
Cristea S, Freyvert Y, Santiago Y, Holmes MC, Urnov FD, Gregory PD, Cost GJ. In vivo cleavage of transgene donors promotes nuclease-mediated targeted integration. Biotechnol Bioeng. 2013;110:871–80.
Lackner DH, Carré A, Guzzardo PM, Banning C, Mangena R, Henley T, et al. A generic strategy for CRISPR-Cas9-mediated gene tagging. Nat. Commun. 2015;6:10237.
Kesavan G, Chekuru A, Machate A, Brand M. CRISPR/Cas9-mediated zebrafish knock-in as a novel strategy to study midbrain-hindbrain boundary development. Front Neuroanat. 2017;11:1–14.
Kimura Y, Hisano Y, Kawahara A, Higashijima S. Efficient generation of knock-in transgenic zebrafish carrying reporter/driver genes by CRISPR/Cas9-mediated genome engineering. Sci Rep. 2014;4:6545.
Shi Z, Wang F, Cui Y, Liu Z, Guo X, Zhang Y, Deng Y, Zhao H, Chen Y. Heritable CRISPR/Cas9-mediated targeted integration in Xenopus tropicalis. FASEB J. 2015;29:4914–23.
Auer TO, Duroure K, De Cian A, Concordet JP, Del Bene F. Highly efficient CRISPR/Cas9-mediated knock-in in zebrafish by homology-independent DNA repair. Genome Res. 2014;24:142–53.
Geisinger JM, Turan S, Hernandez S, Spector LP, Calos MP. In vivo blunt-end cloning through CRISPR/Cas9-facilitated non-homologous end-joining. Nucleic Acids Res. 2016;44:1–15.
Li J, Zhang B, Ren Y, Gu S, Xiang Y, Du J. Intron targeting-mediated and endogenous gene integrity-maintaining knockin in zebrafish using the CRISPR/Cas9 system. Cell Res. 2015;25:634–7.
He X, Tan C, Wang F, Wang Y, Zhou R, Cui D, You W, Zhao H, Ren J, and Feng B. Knock-in of large reporter genes in human cells via CRISPR/Cas9-induced homology-dependent and independent DNA repair. Nucleic Acids Res. 2016; 44:e85.
Brown A, Woods WS, Perez-Pinera P. Multiplexed targeted genome engineering using a universal nuclease-assisted vector integration system. ACS Synth Biol. 2016;5:582–8.
Katoh Y, Michisaka S, Nozaki S, Funabashi T, Hirano T, Takei R, Nakayama K. Practical method for targeted disruption of cilia-related genes by using CRISPR/Cas9-mediated, homology-independent knock-in system. Mol Biol Cell. 2017;28:898–906.
Irion U, Krauss J, Nusslein-Volhard C. Precise and efficient genome editing in zebrafish using the CRISPR/Cas9 system. Development. 2014;141:4827–30.
Wang Y, Cheng X, Shan Q, Zhang Y, Liu J, Gao C, Qiu J-L. Simultaneous editing of three homoeoalleles in hexaploid bread wheat confers heritable resistance to powdery mildew. Nat Biotechnol. 2014;32:947–51.
Yamamoto Y, Bliss J, Gerbi SA. Whole organism genome editing: targeted large DNA insertion via ObLiGaRe nonhomologous end-joining in vivo capture. G3. 2015;5:1843–7.
Guirouilh-Barbat J, Lambert S, Bertrand P, Lopez BS. Is homologous recombination really an error-free process? Front Genet. 2014;5:1–15.
Suzuki K, Tsunekawa Y, Hernandez-Benitez R, Wu J, Zhu J, Kim EJ, et al. In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature. 2016;540:144–9.
Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–21.
Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–7.
Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:1335–8.
Yin H, Song C-Q, Dorkin JR, Zhu LJ, Li Y, Wu Q, et al. Therapeutic genome editing by combined viral and non-viral delivery of CRISPR system components in vivo. Nat Biotechnol. 2016;34:328–33.
Yang Y, Wang L, Bell P, McMenamin D, He Z, White J, et al. A dual AAV system enables the Cas9-mediated correction of a metabolic liver disease in newborn mice. Nat Biotechnol. 2016;34:334–8.
Samulski RJ, Muzyczka N. AAV-mediated gene therapy for research and therapeutic purposes. Annu Rev Virol. 2014;1:427–51.
Wang Z, Zhu T, Qiao C, Zhou L, Wang B, Zhang J, Chen C, Li J, Xiao X. Adeno-associated virus serotype 8 efficiently delivers genes to muscle and heart. Nat Biotechnol. 2005;23:321–8.
Nathwani AC, Reiss UM, Tuddenham EGD, Rosales C, Chowdary P, McIntosh J, et al. Long-term safety and efficacy of factor IX gene therapy in Hemophilia B. N Engl J Med. 2014;371:1994–2004.
McVey M, Lee SE. MMEJ repair of double-strand breaks (director’s cut): deleted sequences and alternative endings. Trends Genet. 2008;24:529–38.
Nakade S, Tsubota T, Sakane Y, Kume S, Sakamoto N, Obara M, et al. Microhomology-mediated end-joining-dependent integration of donor DNA in cells and animals using TALENs and CRISPR/Cas9. Nat Commun. 2014;5:5560.
Sakuma T, Nakade S, Sakane Y, Suzuki K-IT, Yamamoto T. MMEJ-assisted gene knock-in using TALENs and CRISPR-Cas9 with the PITCh systems. Nat Protoc. 2015;11:118–33.
Yao X, Wang X, Liu J, Hu X, Shi L, Shen X, et al. CRISPR/Cas9 – mediated precise targeted integration in vivo using a double cut donor with short homology arms. EBioMedicine. 2017;20:19–26.
Yao X, Wang X, Hu X, Liu Z, Liu J, Zhou H, et al. Homology-mediated end joining-based targeted integration using CRISPR/Cas9. Cell Res. 2017;27:801–14.
Cox DBT, Platt RJ, Zhang F. Therapeutic genome editing: prospects and challenges. Nat Med. 2015;21:121–31.
Swiech L, Heidenreich M, Banerjee A, Habib N, Li Y, Trombetta J, Sur M, Zhang F. In vivo interrogation of gene function in the mammalian brain using CRISPR-Cas9. Nat Biotechnol. 2014;33:102–6.
Long C, Amoasii L, Mireault AA, McAnally JR, Li H, Sanchez-Ortiz E, Bhattacharyya S, Shelton JM, Bassel-Duby R, Olson EN. Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science. 2016;351:400–3.
Soltys S, Rengo G, Rabinowitz JE, Nowak KJ, Duddy W, Partridge T, et al. In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science. 2016;351:403–8.
Li D, Yue Y, Duan D, Mendell JR, Mingozzi F, High KA, et al. In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science. 2016;351:407–12.
Bakondi B, Lv W, Lu B, Jones MK, Tsai Y, Kim KJ, et al. In vivo CRISPR/Cas9 gene editing corrects retinal dystrophy in the S334ter-3 rat model of autosomal dominant retinitis pigmentosa. Mol Ther. 2016;24:556–63.
Luo J, Baranov P, Patel S, Ouyang H, Quach J, Wu F, et al. Human retinal progenitor cell transplantation preserves vision. J Biol Chem. 2014;289:6362–71.
D’Cruz PM, Yasumura D, Weir J, Matthes MT, Abderrahim H, LaVail MM, Vollrath D. Mutation of the receptor tyrosine kinase gene Mertk in the retinal dystrophic RCS rat. Hum Mol Genet. 2000;9:645–51.
Slaymaker IM, Gao L, Zetsche B, Scott DA, Yan WX, Zhang F. Rationally engineered Cas9 nucleases with improved specificity. Science. 2016;351:84–88.
Kleinstiver BP, Pattanayak V, Prew MS, Tsai SQ, Nguyen NT, Zheng Z, Joung JK. High-fidelity CRISPR–Cas9 nucleases with no detectable genome-wide off-target effects. Nature. 2016;529:490–5.
Acknowledgements
We are grateful to Y. Xia and D. O’Keefe for help with manuscript preparation. KS was supported by JSPS KAKENHI (15K21762). JCIB was supported by The Leona M. and Harry B. Helmsley Charitable Trust (2012-PG-MED002), The G. Harold and Leila Y. Mathers Charitable Foundation, NIH (R01HL123755), The McKnight Foundation, The Moxie Foundation, Fundacion Dr. Pedro Guillen, and Universidad Católica San Antonio de Murcia (UCAM).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interests
The authors declare that they have no competing interests.
Rights and permissions
About this article

Cite this article
Suzuki, K., Izpisua Belmonte, J.C. In vivo genome editing via the HITI method as a tool for gene therapy. J Hum Genet 63, 157–164 (2018). https://doi.org/10.1038/s10038-017-0352-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s10038-017-0352-4
This article is cited by
-
Donor template delivery by recombinant adeno-associated virus for the production of knock-in mice
BMC Biology (2024)
-
Precise genome-editing in human diseases: mechanisms, strategies and applications
Signal Transduction and Targeted Therapy (2024)
-
Homology-independent targeted insertion (HITI) enables guided CAR knock-in and efficient clinical scale CAR-T cell manufacturing
Molecular Cancer (2023)
-
Gene therapy for liver diseases — progress and challenges
Nature Reviews Gastroenterology & Hepatology (2023)
-
Antibody-directed extracellular proximity biotinylation reveals that Contactin-1 regulates axo-axonic innervation of axon initial segments
Nature Communications (2023)
