
Abstract
Genome-wide association studies (GWASs) have identified a number of loci for psoriasis but largely ignored non-additive effects. We report a genotypic variability-based GWAS (vGWAS) that can prioritize non-additive loci without requiring prior knowledge of interaction types or interacting factors in two steps, using a mixed model to partition dichotomous phenotypes into an additive component and non-additive environmental residuals on the liability scale and then the Levene’s (Brown–Forsythe) test to assess equality of the residual variances across genotype groups genome widely. The vGWAS identified two genome-wide significant (P < 5.0e−08) non-additive loci HLA-C and IL12B that were also genome-wide significant in an accompanying GWAS in the discovery cohort. Both loci were statistically replicated in vGWAS of an independent cohort with a small sample size. HLA-C and IL12B were reported in moderate gene–gene and/or gene–environment interactions in several occasions. We found a moderate interaction with age-of-onset of psoriasis, which was replicated indirectly. The vGWAS also revealed five suggestive loci (P < 6.76e−05) including FUT2 that was associated with psoriasis with environmental aspects triggered by virus infection and/or metabolic factors. Replication and functional investigation are needed to validate the suggestive vGWAS loci.
Similar content being viewed by others

Introduction
Psoriasis is a chronic inflammatory skin disease mediated by both the innate and adaptive immune systems [1]. Psoriasis is highly heritable with a heritability estimate of 68% (60 to 75%) and a prevalence of around 2.5% among European populations with little gender difference [2, 3]. Genome-wide association studies (GWASs) have highlighted more than 40 loci associated with psoriasis in Caucasian samples, which collectively explain <30% of the total genetic variation [4, 5]. Although meta-analyses of additional GWAS cohorts may reveal additional novel loci, as observed in other immune-mediated diseases, most newly discovered variants are likely to be intergenic with moderate/weak effects and subsequently pose challenges in biological interpretation [6]. One often needs to interrogate GWAS signals in the light of other sources of information such as pathways and regulatory elements [7, 8], where non-additive effects such as gene–gene (GxG) and/or gene–environment (GxE) interactions may also be implicated [9].
Genome-wide exploring GxG and/or GxE interactions in GWAS data has been attempted widely but the results so far have been relatively disappointing mainly due to low power of detection [10,11,12]. Testing for non-additive interactions is more challenging than testing for additive signals, because, for example, detection of a GxG epistatic interaction requires markers to tag the causal variants at both loci and, where that tagging is imperfect, power is adversely affected [12]. A recent genotype–covariate interaction study also showed that GxE interactions jointly could contribute substantially (e.g., 8.1% by genotype–age interactions) to the variation of body mass index but required large samples to detect individually because of likely small effect sizes per interaction [13]. Several studies have reported GxG interactions in psoriasis by examining only the known susceptibility loci such as the major histocompatibility complex (MHC) [14,15,16,17]. A well-known example is the interaction between two GWAS significant loci HLA-C and ERAP1, where ERAP1 encodes an amino peptidase regulating the quality of peptides bound to MHC class I molecules encoded by HLA-C [16, 18]. This epistatic signal is not consistently detected possibly due to the relatively weak interaction effects [19]. Other examples include interactions between the HLA-Cw6 allele and the LCE3C and LCE3B deletion [14, 15, 17], and between HLA-C and IL12B in Chinese populations [17]. These reported GxG interactions each carry a rather moderate association signal, and thus are more useful in indicating regulatory mechanisms than explaining genetic variance. Environmental factors such as smoking and obesity are thought to interact with psoriasis susceptibility genes, but remain to be quantified because these factors are not documented adequately and consistently [20]. Nonetheless, a relatively weak interaction between IL12B and obesity was detected when using large samples [21].
The issue of low power of detection of non-additive loci is likely to be persistent if assuming a specific interaction model (e.g. either GxG or GxE) in a genome search [12, 13]. On the other hand, a locus can be involved in multiple non-additive interactions [22, 23] where the aggregate non-additive effects could be substantial [9]. Genotypic variability-based genome-wide association study (vGWAS) provides an alternative that can prioritize non-additive loci without requiring any prior knowledge of interacting types and factors [12, 24, 25]. vGWAS can achieve this by considering substantial genotypic variability, i.e., differences of phenotypic variation across three SNP genotypes, as the potential interaction signatures tend to be weak when only additive effects are important but strong when non-additive effects such as GxE interactions are important [26, 27]. Additional explicit tests of GxE and/or GxG interactions would be needed but only for the identified vGWAS loci, leading to a power increase attributed to a reduced multiple testing burden [23, 28].
The vGWAS approach has been applied to a number of quantitative traits and revealed interacting loci also carrying major additive effects [23, 28,29,30,31,32]. Recently, we adapted vGWAS to analyze dichotomous disease phenotypes using a two-stage vGWAS approach on the liability scale and identified MHC as a key interacting locus in three sero-positive rheumatoid arthritis (RA) cohorts [22]. These vGWAS findings showed that the MHC genes carry not only important additive but also non-additive effects in RA [22]. In a separate vGWAS meta-analysis of six sero-negative RA cohorts, we identified not only the MHC genes but also novel loci DHCR7 and IRF4 illuminating interesting biology of cutaneous synthesis of vitamin D and light skin color when early humans to adapt to Northern Europe where residents have reduced ultraviolet-B exposure [33]. In addition, a very recent study showed that only variants increasing both the mean and between-subject variance in fasting glucose levels (i.e., with signals in both GWAS and vGWAS) could predict risk of type II diabetes [34].
Nevertheless, there remain some unanswered questions about the vGWAS approach. First, the previous RA vGWAS analyzed cohorts genotyped with immunochip carrying a high-density of markers preselected for a set of immune-relevant genes [35] and consequently provided only a partial genome view of vGWAS. It is therefore of interest to find out whether vGWAS is useful in other diseases and at the genome-wide scale. Second, power of vGWAS is determined by the sample size, coverage and density of genomic markers such as single nucleotide polymorphism (SNP), effect sizes of underlying interactions, and (probably unobserved) interacting factors [22, 26]. Imputation can markedly increase SNP coverage and density, i.e., to increase linkage disequilibrium (LD) with any causal variants [12, 36], but is yet to be employed in vGWAS of diseases. Therefore, we applied the vGWAS approach [22] to analyze two psoriasis cohorts: the Wellcome Trust Case Control Consortium 2 (WTCCC2, for discovery) and the cohort derived for the Collaborative Association Study of Psoriasis (CASP, for replication), to explore these aspects.
Materials and methods
Study cohorts and quality control
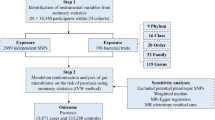
We used the WTCCC2 psoriasis cohort derived as a component of the Wellcome Trust Case Control Consortium 2 project to discover vGWAS signals and the cohort derived for the Collaborative Association Study of Psoriasis (CASP, General Research Use only) to replicate. Both cohorts have been documented in details elsewhere [16, 37]. Briefly, the WTCCC2 cohort samples were recruited from five centers in England, Scotland and Ireland, where phenotypic data and blood samples were collected after research ethics approval was received from each participating institution and after subjects had given written informed consent, and DNA samples were genotyped on the Illumina Human660W-Quad and Illumina custom Human1.2M-Duo for cases and controls respectively. The CASP cohort samples were recruited from the US and all participants provided written informed consent and were genotyped on the PERLEGEN-600K. All experiments (e.g., genotyping) were performed in accordance with relevant guidelines and regulations.
We excluded SNPs on sex chromosomes and samples of non-European origin. The WTCCC2 and CASP datasets had 513,278 and 372,025 SNPs available for imputation, respectively. The two cohorts were pre-phased using SHAPEIT2 [38] and imputed to the 1000 Genomes Project reference panel (phase1 integrated variant set v3) using IMPUTE2 [39]. SNPs with an imputation INFO score <0.8 and indels were excluded and the remaining imputed data were converted to PLINK format. We used PLINK [40] to perform quality control in the imputed as well as the raw data of each cohort to ensure: minor allele frequency >0.01, SNP call rate >0.95, sample call rate >0.99, deviation from Hardy–Weinberg equilibrium P < 1.0e−04.
Two-stage vGWAS
We conducted vGWAS of the raw and imputed data for each cohort using the two-stage approach previously developed for case–control disease phenotypes [22]. Briefly, GCTA [41] was used to compute the genomic relationship matrix (GRM) and subsequently the first ten principal components (PCs) and then to partition the dichotomous phenotypes into polygenic liability risk and residuals for each unrelated individual, where the disease prevalence was set as 0.025, the GRM relatedness threshold was set as 0.05 and the first 10 PCs were fitted as covariates in the mixed model. Then the resultant residuals were tested for variance heterogeneity across three SNP genotypes using the Levene’s (Brown–Forsythe) test implemented in an R package VariABEL [42] that requires no assumption of normally distributed phenotypes:
where the residuals were the trait y; Zi = |yi−ỹgi| is the deviation of y of the ith sample (yi) and the median of y in samples with genotype g (ỹgi); N is the sample size and k is the total possible genotypes; nj is the number of samples with genotype j; Zj. is the mean deviation from the median for genotype j and Z.. is the mean deviation from the overall median. When N is large, T2 is an approximate χ2 test taking two degrees of freedom.
For simplicity, we adopted the GWAS consensus threshold of 5.0e−08 as the genome-wide significance threshold for vGWAS. We also derived the 5% genome-wide suggestive threshold as 6.7e−05 using permutation of the WTCCC2 data in these steps: (a) randomly permute the environmental residuals of samples; (b) perform the Levene’s (Brown–Forsythe) test genome-wide; (c) record the lowest P-value; (d) reiterate steps (a) to (c) 1000 times, sort the recorded P-values and derive the 5% threshold. We considered vGWAS signals exceeding the genome-wide suggestive threshold in WTCCC2 as discoveries and examined their replication in the CASP cohort: as direct replication if the same SNP had two degrees of freedom in the CASP vGWAS with a P-value <5.0e−02, or indirect replication if the SNP is not available but a proxy had a P-value <5.0e−02 and with two degrees of freedom. We reported mostly the results from the imputed data in the main text.
Additional statistical analysis
For comparison we also performed a conventional GWAS for each cohort using PLINK following the previous study example [22]. We used the following logistic regression model to test GxE or GxG interactions for an identified vGWAS SNP K:
where P is the probability of an individual being a case rather than a control in a population, µ is the model constant, β is the effects of fixed covariates (e.g., gender and age-of-onset), βk is the effect of a vGWAS SNP K, βf is the effect of the interacting factor F that can be an environmental factor (i.e., testing GxE) or another SNP (i.e., testing GxG), βkf is effect of interactions between K and F, e is the random error. We quoted SNP genomic locations in the GRCh38/hg38 version throughout.
Results
After quality control and a removal of genomic related individuals based on the imputed data, 2175 cases and 5144 controls each with genotypes of about 5.5 million SNPs were included in the WTCCC2 analyses, in contrast to 849 cases and 641 controls with about 4.6 million SNPs in the CASP analyses (Table 1). The polygenic heritability estimate was 53.7% in WTCCC2, much lower than 67.9% estimated from the CASP cohort that had fewer controls (i.e., possibly inflated genetic effects) but a much larger standard error estimate. On the basis of the GWAS consensus genome-wide significance threshold of 5.0e−08 and a genome-wide suggestive threshold of 6.76e−05 derived from permutation, we found 6298 SNPs in WTCCC2 with only 1% located outside of MHC in contrast to 905 significant SNPs detected in CASP all within MHC (Table 1).
In the discovery cohort WTCCC2, vGWAS identified two genome-wide significant (P < 5.0e−08) loci MHC and IL12B that were also genome-wide significant in the conventional GWAS. The vGWAS also identified suggestive loci KCNH7, ANKRD55, EXOC2, SFTPD, and FUT2 of which only KCNH7 was significant in the GWAS (Fig. 1). On the other hand, a number of genome-wide significant (P < 5.0e−08) GWAS hits, e.g., rs27524 (mapping to ERAP1), were not detected in the vGWAS analysis (Supplementary Table S1). The vGWAS SNPs above the genome-wide suggestive threshold in WTCCC2 were tested in CASP but only the two genome-wide significant SNPs rs115903802 (mapping to HLA-C) and rs4921482 (mapping to IL12B) were replicated (Table 2).

Aligned Manhattan plots of GWAS (left) and vGWAS (right) analyses in WTCCC2 with imputed data. P-values are at the −log10 scale; red line represents the genome-wide significance threshold; blue line represents the genome-wide suggestive threshold for vGWAS derived from permutation; significant loci are annotated to genes in red (or blue if reached only the vGWAS suggestive threshold, with the minimum P-value in brackets if <1.0e−20)
Imputation increased both the strength and number of vGWAS signals in the WTCCC2 cohort, where rs115903802 (HLA-C), rs4921482 (IL12B), and rs601338 (FUT2) were all imputed SNPs each carrying a stronger signal than that from the corresponding genotyped SNPs within the gene region (Table 2; Supplementary Fig. S1; Supplementary Table S2). In addition, most of the identified genotyped vGWAS SNPs (Supplementary Table S2) gained certain signal strength from imputation that enabled a better estimate/partition of the additive/non-additive genetic variances, e.g., the heritability estimate was 0.495 in the raw data without imputation, much lower than 0.537 estimated from the imputed data (Table 1).
We tested the genome-wide significant vGWAS SNPs for GxE interactions with either gender or age-of-onset in WTCCC2. We found a moderate interaction (interaction P = 8.1e−05, with the full two degrees of freedom (df)) between age-of-onset and rs3132560 (vGWAS P-value of 5.4e−18) mapping to the PSORS1C1 gene. The age-of-onset interaction was not directly replicated (interaction P = 9.1e−02, with 2 df) but indirectly by a proxy SNP rs1265092 (interaction P = 3.2e−04, with 2 df) in CASP. We also tested GxG interactions within the MHC region and found signals similar to previously reported [43] (results not showed). In addition, we explicitly tested the reported interaction between HLA-C and ERAP1 but found no important signal in either cohort.
Discussion
We present a genome-wide profile of vGWAS of psoriasis for the first time. Similar to our previous vGWAS of RA based on immunochip data [22], vGWAS was able to identify non-additive loci that were either significant in the conventional GWAS (e.g., HLA-C, IL12B), or not significant in the GWAS but detected in previous GWAS meta-analyses, i.e., EXCO2 and ANKRD55 for psoriasis [5, 44] and SFTPD for RA [45]. In addition, vGWAS of WTCCC2 identified a suggestive locus FUT2 that was only claimed genome-wide significant very recently in a large scale GWAS meta-analysis combining WTCCC2 with seven other Caucasian cohorts [46]. Only the two major psoriasis loci HLA-C and IL12B were replicated in the CASP cohort, which might be viewed as a limitation of the study (Table 2). However, it should be noted that sample sizes in CASP were much smaller than WTCCC2 (Table 1), limiting the power to detect modest effect sizes. Also limited by the low power issue, we only found evidence of GxG and GxE in MHC region but not in IL12B. Nevertheless, both HLA-C and IL12B were previously known interacting loci [14,15,16,17, 21], suggesting vGWAS does pick up non-additive signals correctly.
As expected, imputation was found to be important to increase power of detection in vGWAS [12, 22]. The increase was seen in at least three aspects: (a) better estimates of additive and non-additive variances, and hence better phenotypes for vGWAS, (b) stronger vGWAS signals in known loci (e.g., HLA-C and IL12B) because of imputed SNPs better tagging causal variants, (c) for the same reason, improving the chances of identifying novel associations, e.g., without imputation FUT2 would have been missed in several studies [46, 47]. Nonetheless, additional cares are recommended when interpreting results derived from imputed data as imputation itself could have posed extra uncertainty to genotypic variability. Further replication tests and functional investigation of FUT2 and the other suggestive vGWAS loci in the relevant environmental context are therefore recommended.
FUT2 encodes alpha-(1,2)fucosyltransferase which regulates the H antigen (precursor of the human ABO blood group antigens) on the surface of epithelial cells and in body fluids. Interestingly, FUT2 was found moderately associated with psoriasis in a cross-disease GWAS combining psoriasis with cardiovascular and metabolic diseases, where rs492602 (in perfect LD with the vGWAS SNP rs681343) had a GWAS P-value of 6.59e−05 in the WTCCC2 cohort [47]. In another early cross-disease GWAS combining samples of Crohn disease and psoriasis, the genome-wide significant SNP rs281379 mapping to FUT2, had a nearly genome-wide significant association with psoriasis when combining all psoriasis samples (P = 7.86e−08) [48]. These cross-disease GWAS results suggested that FUT2 perhaps carried only moderate additive effects in association with psoriasis. Such moderate associations thus require large samples to identify as showed in the latest large GWAS meta-analysis of psoriasis in Caucasians where rs492602 was the lead SNP.
From this perspective, vGWAS has potential in discovering novel associations driven by both non-additive and additive effects such as DHCR7 and IRF4 showed previously [33] and FUT2 here. There is evidence supporting that FUT2 could be important in responding to different environmental factors. For example, a separate study of FUT2 in Behçet’s disease [49] highlighted environmental aspects (e.g., infection by virus, inflammation by rheumatic, or metabolic factors) in the heterogeneity of the FUT2 associations. Such environmental aspects may reduce the power of association tests based solely on additive effects. Similarly, the vGWAS locus EXCO2, known to be responsible for skin color and highly related to sunlight, was only detected in GWAS of psoriasis with very large samples due to moderate additive effects, or in GWAS of a directly linked phenotype (e.g., tanning phenotype, cutaneous basal cell carcinoma) in samples with European ancestry with sufficient allele frequencies [50, 51].
vGWAS appears to be an effective filter to improve power of detection GxG interactions by concerning only the identified vGWAS loci [12, 22, 33]. However, other than the known GxG interactions within the MHC region [33, 43, 52], this study is obviously low powered to explicitly test either GxG or GxE interactions at a given model because of small samples in each single GWAS cohort. This is in line with the previous observations where multiple reasons could account for lack of robust findings of interactions, e.g., unmeasured causal environmental triggers [12, 13, 22, 33]. These reasons are also relevant to explain limited replication of the identified vGWAS loci and thus warrant further efforts to dissect any interactions potentially involved these loci. Our results together indicate that vGWAS is a useful complement to GWAS in discovering loci where non-additive effects are equally (if not more) important but requires large samples.
Change history
23 May 2019
This article was originally published under a CC BY-NC-SA License, but has now been made available under a CC BY 4.0 License.
References
Lowes MA, Suarez-Farinas M, Krueger JG. Immunology of psoriasis. Annu Rev Immunol. 2014;32:227–55.
Chandran V, Raychaudhuri SP. Geoepidemiology and environmental factors of psoriasis and psoriatic arthritis. J Autoimmun. 2010;34:J314–21.
Lonnberg AS, Skov L, Skytthe A, Kyvik KO, Pedersen OB, Thomsen SF. Heritability of psoriasis in a large twin sample. Br J Dermatol. 2013;169:412–6.
Tsoi LC, Spain SL, Ellinghaus E, Stuart PE, Capon F, Knight J, et al. Enhanced meta-analysis and replication studies identify five new psoriasis susceptibility loci. Nat Commun. 2015;6:7001.
Tsoi LC, Spain SL, Knight J, Ellinghaus E, Stuart PE, Capon F, et al. Identification of 15 new psoriasis susceptibility loci highlights the role of innate immunity. Nat Genet. 2012;44:1341–8.
Diogo D, Okada Y, Plenge RM. Genome-wide association studies to advance our understanding of critical cell types and pathways in rheumatoid arthritis: recent findings and challenges. Curr Opin Rheumatol. 2014;26:85–92.
Parkes M, Cortes A, van Heel DA, Brown MA. Genetic insights into common pathways and complex relationships among immune-mediated diseases. Nat Rev Genet. 2013;14:661–73.
Pers TH, Karjalainen JM, Chan Y, Westra HJ, Wood AR, Yang J, et al. Biological interpretation of genome-wide association studies using predicted gene functions. Nat Commun. 2015;6:5890.
Zuk O, Hechter E, Sunyaev SR, Lander ES. The mystery of missing heritability: genetic interactions create phantom heritability. Proc Natl Acad Sci USA. 2012;109:1193–8.
Lehner B. Genotype to phenotype: lessons from model organisms for human genetics. Nat Rev Genet. 2013;14:168–78.
Thomas D. Gene–environment-wide association studies: emerging approaches. Nat Rev Genet. 2010;11:259–72.
Wei W-H, Hemani G, Haley CS. Detecting epistasis in human complex traits. Nat Rev Genet. 2014;15:722–33.
Robinson MR, English G, Moser G, Lloyd-Jones LR, Triplett MA, Zhu Z, et al. Genotype–covariate interaction effects and the heritability of adult body mass index. Nat Genet. 2017;49:1174–81.
de Cid R, Riveira-Munoz E, Zeeuwen PL, Robarge J, Liao W, Dannhauser EN, et al. Deletion of the late cornified envelope LCE3B and LCE3C genes as a susceptibility factor for psoriasis. Nat Genet. 2009;41:211–5.
Riveira-Munoz E, He SM, Escaramis G, Stuart PE, Huffmeier U, Lee C, et al. Meta-analysis confirms the LCE3C_LCE3B deletion as a risk factor for psoriasis in several ethnic groups and finds interaction with HLA-Cw6. J Invest Dermatol. 2011;131:1105–9.
Strange A, Capon F, Spencer CC, Knight J, Weale ME, Allen MH, et al. A genome-wide association study identifies new psoriasis susceptibility loci and an interaction between HLA-C and ERAP1. Nat Genet. 2010;42:985–90.
Zheng HF, Zuo XB, Lu WS, Li Y, Cheng H, Zhu KJ, et al. Variants in MHC, LCE and IL12B have epistatic effects on psoriasis risk in Chinese population. J Dermatol Sci. 2011;61:124–8.
Yin XY, Zhang R, Cheng H, Pan Q, Shen CB, Fan X, et al. Gene–gene interactions between HLA-C, ERAP1, TNFAIP3 and TRAF3IP2 and the risk of psoriasis in the Chinese Han population. Br J Dermatol. 2013;169:941–3.
Lysell J, Padyukov L, Kockum I, Nikamo P, Stahle M. Genetic association with ERAP1 in psoriasis is confined to disease onset after puberty and not dependent on HLA-C*06. J Invest Dermatol. 2013;133:411–7.
Naldi L. Risk factors for psoriasis. Curr Derm Rep. 2013;2:58–65.
Li WQ, Han JL, Zhang MF, Qureshi AA. Interactions between adiposity and genetic polymorphisms on the risk of psoriasis. Br J Dermatol. 2013;168:639–42.
Wei WH, Bowes J, Plant D, Viatte S, Yarwood A, Massey J, et al. Major histocompatibility complex harbors widespread genotypic variability of non-additive risk of rheumatoid arthritis including epistasis. Sci Rep. 2016;6:25014.
Brown AA, Buil A, Vinuela A, Lappalainen T, Zheng HF, Richards JB, et al. Genetic interactions affecting human gene expression identified by variance association mapping. eLife. 2014;3:e01381.
Falconer DS. Selection in different environments: effects on environmental sensitivity (reaction norm) and on mean performance. Genet Res. 1990;56:57–70.
Visscher PM, Posthuma D. Statistical power to detect genetic Loci affecting environmental sensitivity. Behav Genet. 2010;40:728–33.
Struchalin MV, Dehghan A, Witteman JC, van Duijn C, Aulchenko YS. Variance heterogeneity analysis for detection of potentially interacting genetic loci: method and its limitations. BMC Genet. 2010;11:92.
Ronnegard L, Valdar W. Recent developments in statistical methods for detecting genetic loci affecting phenotypic variability. BMC Genet. 2012;13:63.
Pare G, Cook NR, Ridker PM, Chasman DI. On the use of variance per genotype as a tool to identify quantitative trait interaction effects: a report from the Women’s Genome Health Study. PLoS Genet. 2010;6:e1000981.
Yang J, Loos RJF, Powell JE, Medland SE, Speliotes EK, Chasman DI, et al. FTO genotype is associated with phenotypic variability of body mass index. Nature. 2012;490:267–72.
Topless R, Flynn T, Cadzow M, Stamp L, Dalbeth N, Black M, et al. Association of SLC2A9 genotype with phenotypic variability of serum urate in pre-menopausal women. Front Genet. 2015;6:313.
Hulse AM, Cai JJ. Genetic variants contribute to gene expression variability in humans. Genetics. 2013;193:95–108.
Wang G, Yang E, Brinkmeyer-Langford CL, Cai JJ. Additive, epistatic, and environmental effects through the lens of expression variability QTL in a twin cohort. Genetics. 2014;196:413–25.
Wei WH, Viatte S, Merriman TR, Barton A, Worthington J. Genotypic variability based association identifies novel non-additive loci DHCR7 and IRF4 in sero-negative rheumatoid arthritis. Sci Rep. 2017;7:5261.
Ivarsdottir EV, Steinthorsdottir V, Daneshpour MS, Thorleifsson G, Sulem P, Holm H, et al. Effect of sequence variants on variance in glucose levels predicts type 2 diabetes risk and accounts for heritability. Nat Genet. 2017;49:1398–402.
Cortes A, Brown MA. Promise and pitfalls of the immunochip. Arthritis Res Ther. 2011;13:101.
Howey R, Cordell HJ. Imputation without doing imputation: a new method for the detection of non-genotyped causal variants. Genet Epidemiol. 2014;38:173–90.
Nair RP, Duffin KC, Helms C, Ding J, Stuart PE, Goldgar D, et al. Genome-wide scan reveals association of psoriasis with IL-23 and NF-kappaB pathways. Nat Genet. 2009;41:199–204.
Delaneau O, Zagury JF, Marchini J. Improved whole-chromosome phasing for disease and population genetic studies. Nat Methods. 2013;10:5–6.
Howie BN, Donnelly P, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 2009;5:e1000529.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75.
Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet. 2011;88:76–82.
Struchalin MV, Amin N, Eilers PH, van Duijn CM, Aulchenko YS. An R package “VariABEL” for genome-wide searching of potentially interacting loci by testing genotypic variance heterogeneity. BMC Genet. 2012;13:4.
Wu X, Dong H, Luo L, Zhu Y, Peng G, Reveille JD, et al. A novel statistic for genome-wide interaction analysis. PLoS Genet. 2010;6:e1001131.
Stein MB, Chen CY, Ursano RJ, Cai T, Gelernter J, Heeringa SG, et al. Genome-wide association studies of posttraumatic stress disorder in 2 cohorts of US army soldiers. JAMA Psychiatry. 2016;73:695–704.
Okada Y, Wu D, Trynka G, Raj T, Terao C, Ikari K, et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature. 2014;506:376–81.
Tsoi LC, Stuart PE, Tian C, Gudjonsson JE, Das S, Zawistowski M, et al. Large scale meta-analysis characterizes genetic architecture for common psoriasis associated variants. Nat Commun. 2017;8:15382.
Lu Y, Chen H, Nikamo P, Qi Low H, Helms C, Seielstad M, et al. Association of cardiovascular and metabolic disease genes with psoriasis. J Invest Dermatol. 2013;133:836–9.
Ellinghaus D, Ellinghaus E, Nair RP, Stuart PE, Esko T, Metspalu A, et al. Combined analysis of genome-wide association studies for Crohn disease and psoriasis identifies seven shared susceptibility loci. Am J Hum Genet. 2012;90:636–47.
Xavier JM, Shahram F, Sousa I, Davatchi F, Matos M, Abdollahi BS, et al. FUT2: filling the gap between genes and environment in Behçet’s disease? Ann Rheum Dis 2013; 74:618–24.
Nan H, Kraft P, Qureshi AA, Guo Q, Chen C, Hankinson SE, et al. Genome-wide association study of tanning phenotype in a population of European ancestry. J Invest Dermatol. 2009;129:2250–7.
Nan H, Xu M, Kraft P, Qureshi AA, Chen C, Guo Q, et al. Genome-wide association study identifies novel alleles associated with risk of cutaneous basal cell carcinoma and squamous cell carcinoma. Hum Mol Genet. 2011;20:3718–24.
Wei WH, Loh CY, Worthington J, Eyre S. Immunochip analyses of epistasis in rheumatoid arthritis confirm multiple interactions within MHC and suggest novel non-MHC epistatic signals. J Rheumatol. 2016;43:839–45.
Acknowledgements
WHW is funded by the Higher Education Funding Council for England (HEFCE) previously and currently University of Otago and Cure Kids NZ. We acknowledge the assistance given by IT Services and the use of the Computational Shared Facility at The University of Manchester. The funding of the Wellcome Trust Case Control Consortium 2 project (083948/Z/07/Z) was provided by the Wellcome Trust. We also thank S. Bertrand, J. Bryant, SL Clark, JS Conquer, T. Dibling, J.C. Eldred, S. Gamble, C. Hind, A. Wilk, C.R. Stribling, and S. Taylor of the Wellcome Trust Sanger Institute’s Sample and Genotyping Facilities for technical assistance. Funding for the Collaborative Association Study of Psoriasis was provided by the National Institutes of Health, the Foundation for the National Institutes of Health, and the National Psoriasis Foundation. Support for genotyping of samples was provided through the Genetic Association Information Network (GAIN). The dataset(s) used for the analyses described in this manuscript were obtained from the database of Genotypes and Phenotypes (dbGaP) found at http://www.ncbi.nlm.nih.gov/gap through dbGaP accession number phs000019 (project ID 7658). Samples and associated phenotype data for the Collaborative Association Study of Psoriasis were provided by Drs. James T Elder (University of Michigan, Ann Arbor, MI), Gerald G Krueger (University of Utah, Salt Lake City, UT), Anne Bowcock (Washington University, St. Louis, MO), and Gonçalo R Abecasis (University of Michigan, Ann Arbor, MI). For a description of the dataset, phenotypes, genotype data, and quality control procedures see Nair et al. [37]
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Ethical statement
We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wei, WH., Massey, J., Worthington, J. et al. Genotypic variability-based genome-wide association study identifies non-additive loci HLA-C and IL12B for psoriasis. J Hum Genet 63, 289–296 (2018). https://doi.org/10.1038/s10038-017-0350-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s10038-017-0350-6
This article is cited by
-
Disentangling the complexity of psoriasis in the post-genome-wide association era
Genes & Immunity (2023)
-
Discovering Genetic Factors for psoriasis through exhaustively searching for significant second order SNP-SNP interactions
Scientific Reports (2018)
