
Abstract
Periventricular leukomalacia (PVL) in the premature infant represents the major substrate underlying cognitive deficits and cerebral palsy and is characterized as focal periventricular necrosis and diffuse gliosis in the immature cerebral white matter. We have recently shown a significant decrease in the density of neurons in PVL relative to controls throughout the white matter, including the subventricular, periventricular, and subcortical regions. These neurons are likely to be remnants of the subplate and/or GABAergic neurons in late migration to the cerebral cortex, both of which are important for proper cortical circuitry in development and throughout adulthood. Here, we tested the hypothesis that intrinsic repair occurs in PVL to attempt to compensate for the deficits in white matter neurons. By using doublecortin (DCX) immunopositivity as a marker of postmitotic migrating neurons, we found significantly increased densities (p < 0.05) of DCX-immunopositive cells in PVL cases (n = 9) compared with controls (n = 7) in the subventricular zone (their presumed site of origin), necrotic foci, and subcortical white matter in the perinatal time-window, i.e. 35–42 postconceptional weeks. These data provide the first evidence suggestive of an attempt at neuronal repair or regeneration in human neonatal white matter injury.
Similar content being viewed by others
Main
A major challenge faced by modern neonatology is the prevention of neurological sequelae in premature infants who survive the neonatal period. In the United States alone, ∼63,000 infants are born prematurely (birth weights < 1,500 g) and 350,000 are born late preterm (34–36 gestational wk) (1,2). Although the overall survival rate of all premature groups has sharply increased to >90% (3), the burden of neurological disability remains great: 25–50% of the low birth weight infants born each year develop major cognitive deficits, 10% develop cerebral palsy (1), and 2.1% of late preterm infants develop variable cognitive disabilities, cerebral palsy, and/or seizures, a 2-fold increase compared with term infants (2). The encephalopathy of prematurity (EP) is the major neuropathologic substrate of neurological deficits in preterm survivors. It involves variable combinations of white matter damage in the form of periventricular leukomalacia (PVL) and diffuse white matter gliosis, gray matter damage (neuronal necrosis, neuronal loss, and/or gliosis), and focal and diffuse axonal injury in the cerebral white matter (1,4,5). The pathogenesis of EP reflects cerebral ischemia/reperfusion in the critically ill premature infant with maternal-fetal infection or inflammation contributing to the damage in some cases (1,4,6,7). Recently, we have shown evidence supporting the understudied possibility that “bystander” injury to white matter neurons, i.e. late-migrating GABAergic neurons and/or residual subplate neurons, in human PVL is a critical part of EP that similarly contributes to impaired cognition in preterm survivors (Andiman S.E. et al, Loss of white matter neurons in periventricular leukomalacia; implications for cognitive deficits in survivors of prematurity, Society for Neuroscience, October 17–21, 2009, Chicago, IL, Abstract 736.26). In this study, we examine the innate capacity of the perinatal brain for neuronal repair or regeneration of these white matter neurons. Although human data in adult neurological disorders such as Alzheimer disease (8) and multiple sclerosis (9) and experimental data from perinatal animal models indicate that spontaneous repair occurs (10–15), data obtained directly from study of the developing human brain are rare (16–18). In PVL, stem cell markers have been used in only two studies to show increased nestin expression in glia and neurons. This increased expression was attributed to nestin up-regulation in response to injury rather than regeneration of new cells (16,17). In this study, we focused on the possibility that the deficit in white matter neurons in PVL is replenished by newly generated neurons that migrate into the regions of the focally necrotic lesions (most severe damage) and/or the surrounding gliotic white matter (less severe damage) from their site of generation in the subventricular zone (SVZ).
We hypothesized that the density of migrating white matter neurons is increased in the SVZ, periventricular necrotic foci, and/or adjacent white matter in PVL. We applied the immunomarker doublecortin (DCX), a microtubule-associated protein localized to postmitotic migrating neurons (19,20), which has been used as an indicator of neurogenesis in two adult human and multiple rodent studies (10–15,21,22).
MATERIALS AND METHODS
Clinical database.
Archival paraffin-embedded tissue was collected from the autopsy services at Children's Hospital Boston and Brigham and Women's Hospital. Parental permission for research was given according to the guidelines of the Human Study Protection Committees at our hospitals. The neuropathology in all cases was assessed by standard histological analysis, including the use of conventional staining with hematoxylin and eosin (H&E)/Luxol fast blue (counter stain for myelin) and immunostaining for glial fibrillary acidic protein (GFAP), a marker of reactive astrocytes. PVL was defined as histopathology in the immature cerebral white matter with: 1) necrotic foci in the periventricular and/or central white matter and 2) diffuse astrogliosis and microglial activation in the surrounding white matter (23). Blocks were selected for DCX analysis in PVL based on the presence of one or more focally necrotic lesions. We subclassified PVL into three temporal stages, acute, organizing or subacute, and chronic as previously described (23). All but one PVL case were at organizing stage with the remaining case being chronic. The control cases were defined as perinatal deaths in which neuropathologic examination revealed no or minimal neuropathologic changes including PVL. The autopsy reports were reviewed for major clinical findings, systemic autopsy diagnoses, and neuropathologic findings (Table 1).
Single-label immunocytochemistry in paraformaldehyde-fixed, paraffin-embedded tissue.
For DCX immunocytochemistry, a rabbit polyclonal antibody made against the C-terminus of human DCX was used at a concentration of 1:200 (Abcam, Cambridge, MA). A polyclonal antibody to Ki67 (1:200; Abcam) was used to examine cellular proliferation on sections adjacent to those labeled with DCX. Standard methods in deparaffinized tissue sections (5 μm) were performed (24). Negative controls omitted the primary antibody.
Double-label immunocytochemistry in paraformaldehyde-fixed, paraffin-embedded tissue.
Tissue was processed as mentioned above, and sections were incubated with DCX (rabbit polyclonal; 1:200; Abcam) and Tuj1 (monoclonal; 1:1500; Covance, Emeryville, CA), Map2 (SMI 52 monoclonal; 1:800; Covance), or GFAP (SMI 26 monoclonal; 1:1000; Covance). Fluorescence was detected with Alexa Fluor goat anti-rabbit 488 and Alexa Fluor goat anti-mouse 594 (1:1000; Molecular Probes, Eugene, OR). For double label of DCX with Olig2 and GAD67/65, a goat polyclonal antibody to DCX (1:100; Santa Cruz, Santa Cruz, CA) was incubated with either a rabbit polyclonal for Olig2 (1:200; Millipore, Billerica, MA) or a rabbit polyclonal for GAD67/65 (1:1000; Abcam) followed by detection with Alexa Fluor donkey anti-rabbit 594 and Alexa Fluor donkey anti-goat 488 (1:1000; Molecular Probes). For double label of DCX and tomato lectin, rabbit polyclonal DCX (1:200); Abcam) was incubated with a biotin-conjugated tomato lectin (1:200; Vector Laboratories, Burlingame, CA) followed by incubation in Alexa Fluor goat anti-rabbit 488 (1:1000; Molecular Probes) and Steptavidin-594 (1:200; Molecular Probes). Negative controls omitted the primary antibodies.
Quantitation of DCX density.
To quantify DCX density in PVL and control tissue sections, the Neurolucida software program was used with a motorized x-y stage (MicroBrightField, VT). The spatial distribution of DCX was determined with two-dimensional graphic plots. At low magnification (4×), the boundaries of the cerebral cortex, white matter, ventricular border, and necrotic periventricular foci were outlined. To quantitate the density of DCX cells in the SVZ and white matter, a grid system was used as previously published by us (24). For PVL cases, one box, 4 mm2, was placed in horizontal alignment with the SVZ; a 4-mm2 box was placed directly over the focal necrosis of PVL; and a 4-mm2 box was placed adjacent to the box containing the focal necrosis in the diffusely gliotic component of PVL. For control cases, a 4-mm2 box was placed in alignment with the SVZ; two 4-mm2 boxes were placed in a distribution equivalent to the boxes containing the focal necrotic and diffusely gliotic components of PVL. An average of both of the white matter boxes in controls were used for comparison with the PVL focal necrotic box and separately with the PVL diffusely gliotic box. For DCX density in the cortex, three separate boxes (each an average of 750 μm in width) were drawn in cortical areas overlying the focal necrosis in PVL cases and in equivalent areas in the control cases. For subcortical DCX density, the subcortical region 750 μm beneath layer VI was demarcated along the border of the cortex overlying the focal necrosis in PVL and in an equivalent region in control cases. The density of DCX+ cells was calculated by dividing the number of cells in the grid or subcortical region by the area and expressed as cells per square millimeter. Cell counting was performed at 40× magnification. Ki67 density measurements in the SVZ were taken from a 4-mm2 area identical to that used for the DCX density. To quantitate the fluorescent double-label immunocytochemistry, we were unable to use the above grid system due to a lack of fluorescent capability equipped with Neurolucida software; therefore, the percent of DCX+ cells colocalizing with either MAP2, NeuN, or GAD67/65 was determined in 3–5 high power fields (40×) within the white matter.
Statistical analysis.
Wilcoxon rank sum tests were used to compare PVL and control cases. No adjustments were made for GA, postnatal age, or postmortem interval as initial analysis of covariance models showed no effects. Similarly, there was no difference in DCX density data across different brain regions; thus, data from all regions were combined for statistical analyses. Fisher exact tests were used for the analysis of categorical clinicopathological data (Table 1).
RESULTS
We analyzed DCX in nine PVL cases and seven controls without PVL in the perinatal period of 35–42 postconceptional weeks. Clinicopathologic information on all cases is listed in Table 1. Although PVL cases tended to be born earlier (p = 0.05) and live significantly longer (p = 0.03) than controls, there was no difference in the postconceptional age between the two groups (p = 0.91; Table 1). Postmortem intervals in PVL cases tended to be on average 7 h shorter than control cases (p = 0.08). There was no difference between PVL cases and controls in the incidence of congenital heart disease (p = 0.61), necrotizing enterocolitis (p = 0.44), sepsis (p = 0.21), or germinal matrix hemorrhages (p = 0.44). The congenital heart disease in the PVL cases and controls was complex, severe, and complicated by cyanosis and need for vasoactive drugs. It included hypoplastic left heart syndrome (n = 2 controls), transposition of the great vessels with hypoplastic aortic arch (n = 1 PVL case), coarctation of the aorta (n = 1 PVL case and n = 1 control), biventricular hypertrophy (n = 1 PVL case), and combined ventricular and atrial septal defects (n = 1 control). All PVL cases and controls required cardiopulmonary resuscitation of variable periods at some point in their hospital course and all received vasoactive drugs. Status epilepticus was not present in either of the two PVL cases with a seizure disorder. The germinal matrix hemorrhage in the control case was minimal and subependymal (grade I). Sections of parietal (n = 5, PVL and n = 2, control), occipital (n = 1, PVL and n = 1, control), posterior frontal (n = 3, PVL and n = 3, control), or temporal (n = 0, PVL and n = 1, control) were examined.
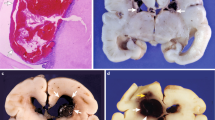
Of the nine PVL cases, four (44%) had gross cystic lesions at autopsy (Fig. 1A), whereas the remainder had necrotic foci only identified microscopically (1–2 mm in diameter). Cells positive for DCX were identified microscopically within (Fig. 1B and C) and adjacent to the periventricular foci of necrosis (Fig. 1D and E) and within the diffusely gliotic component surrounding the necrosis (Fig. 1F). Morphologically, these cells appeared to be migrating neurons with a small round soma (4–11 μm in diameter) and a single, delicate trailing process (Fig. 1C, E, and F). In the SVZ, DCX+ cells were small (3–8 μm in diameter) with scant cytoplasm and trailing processes of variable lengths. Within the SVZ, DCX colocalized with the immature neuronal marker Tuj1 (beta-tubulin III; Fig. 2A). Double-label immunocytochemistry with mature neuronal markers showed that only a small subpopulation of DCX+ cells colocalized with MAP2 (1.1 ± 1.4%; Fig. 2B) and NeuN (2.4 ± 2.6%; Fig. 2C) within the white matter, suggesting a more advanced maturation state in only a small portion of the cells. A subpopulation (35.3 ± 8.3%) of DCX+ cells colocalized with GAD67/65, consistent with the differentiation of these migrating DCX+ cells into a GABAergic phenotype (Fig. 2D). Within the white matter, DCX did not colocalize with the Olig2 antibody for the oligodendrocytic lineage (Fig. 2E), GFAP for the astrocytic lineage (Fig. 2F), and tomato lectin for the macrophage or microglia lineage (Fig. 2G).

The pathologic features of PVL. (A) The macroscopic appearance of PVL in a neonatal brain at 42 postconceptional weeks with bilateral cystic cavities (arrows) in the periventricular white matter in the parietal region at the level of the atrium of the lateral ventricle. (B and D) Microscopic features of PVL in two neonatal brains at 38 and 39 postconceptional weeks. Microcysts (arrowheads) with (D) and without (B) hemorrhage are surrounded by reactive astrocytes (H&E/Luxol fast blue). The insert boxes indicate the location of the DCX+ cells (arrows) immediately adjacent to the focal necrosis (C and E). DCX+ cells in the diffusely gliotic white matter in a PVL case at 38 postconceptional weeks (F). Absence of DCX+ cells in the white matter of a control case at 38 postconceptional weeks (G).

Double-label immunocytochemistry to confirm neuronal phenotype of DCX cells in the white matter of PVL. (A) DCX colocalizes with the early neuronal marker Tuj1 in the SVZ. (B) There is negligible colocalization of DCX cells with mature neuronal marker MAP2. A small subpopulation of DCX+ cells colocalizes with mature neuronal marker NeuN (C) and GAD67/65, the marker of the GABAergic phenotype (D). DCX does not colocalize with the glial markers Olig2 for OLs (E), GFAP for reactive astrocytes (F), and tomato lectin (TL) for microglia and macrophages (G). All images are at 40×. Scale bar = 30 μm.
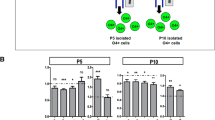
To quantitate the density of DCX+ cells in different subdivisions of the cerebral white matter in PVL cases, we used a computer-based cell counting program (Neurolucida) that allowed for spatial analysis (Fig. 3A) (24). With this method, we found a significant increase in the density of DCX+ cells in PVL cases compared with controls in the following sites: 1) SVZ (p = 0.02); 2) regions in and immediately adjacent to the focally necrotic lesions (p = 0.01); and 3) subcortical white matter directly beneath the cortex (p = 0.02; Fig. 3B). We found a marginally significant increase in regions in the white matter surrounding the necrotic foci (diffusely gliotic lesion; p = 0.06). Within the cortex itself, there was a nonsignificant trend (p = 0.13) toward increased DCX+ cell density in the PVL cases compared with controls (Fig. 3B). To address the question of whether the increased density of DCX+ cells in the SVZ in PVL was a result of increased proliferation, we used reactivity to Ki67, a marker of cellular proliferation. We found no significant increase in PVL cases compared with controls (p = 0.78) in the density of Ki67 cells (Fig. 4). Of note, there were no significant differences in the density of DCX+ or Ki67+ cells in the SVZ or white matter regions between the frontal, parietal, occipital, or temporal lobes (data not shown).

(A) Neurolucida image of a PVL case (posterior frontal region) at 40 postconceptional weeks for the quantitation of DCX density (left). Boundary contours are drawn in the subcortical white matter, the cerebral white matter, and cortex; boxes overlay the subventricular zone, necrotic focus, adjacent white matter (diffuse lesion), and cerebral cortex. The posterior frontal section corresponding to the Neurolucida image and stained with H&E (right). Ven, ventricle; CC, corpus callosum. An asterisk marks the noncystic focal organizing necrosis from which cell counts were taken. This focal necrosis is adjacent to a cystic cavity identified on both the Neurolucida image and the H&E section. (B) DCX density counts in PVL and controls (cells/mm2). Black bars indicate PVL, and white bars indicate controls. The mean and standard errors for each site are as follows: SVZ, PVL = 75.32 ± 21.94, control = 23.86 ± 6.51; focal necrosis, PVL = 11.46 ± 2.52, control = 2.87 ± 1.0; diffuse white matter, PVL = 6.51 ± 1.50, control = 2.87 ± 1.0; cortex, PVL = 8.86 ± 2.91, control = 2.93 ± 1.30; and subcortical, PVL = 4.56 ± 0.82, control = 1.76 ± 0.86. *p value < 0.05.

There is an increase in the density of DCX+ cells in the SVZ of a PVL case at 39 postconceptional weeks (A) compared with a control at 38 postconceptional weeks (B). In the same region of the SVZ on an adjacent section, however, there is little Ki67 expression in either the PVL case (C) or control (D).
DISCUSSION
The major finding of this study is that the human neonatal brain demonstrates the potential capacity for neuronal replenishment in the cerebral white matter in PVL. The significantly increased density of DCX+ cells in the SVZ in proximity to the focally necrotic lesions suggests that the regenerative capacity originates in this germinal site, an idea supported by neonatal animal models of hypoxia-ischemia (10–15) and by reports of human adult brain injury (9,25). With DCX as an immunomarker, animal models of neonatal hypoxia-ischemia show an increased migration of immature neurons from the SVZ to distant sites of injury (10–15), with neurogenesis persisting for even months after the initial insult (10). In animal models, this replenishment is associated with increased cellular proliferation in the SVZ (10–15), a finding that differs from our observation that Ki67 immunostaining is not increased in the SVZ in human PVL. This lack of proliferation in PVL potentially reflects a reservoir of multipotential stem cells and/or neuronal progenitors in the human SVZ that respond to injury by differentiation and migration outward without “additional” proliferation. Alternatively, we may not have captured acute and short-lived SVZ cellular proliferation, as our PVL cases demonstrated more advanced tissue injury, i.e. organizing focal lesions with macrophagocytic infiltration indicative of injury at least 3–5 d old and probably longer (23). Analysis in difficult-to-obtain hyperacute PVL is needed to address this latter possibility. An alternative explanation for our finding of increased DCX+ cells in the white matter in PVL compared with controls is that PVL caused an impairment of cell migration from the germinal zones such that immature neurons (DCX+) are delayed in reaching a site for final maturation. Our increase in DCX+ cells in the white matter could potentially reflect the inclusion of these delayed migrators in our cell counts. This distinction requires further research. An unavoidable limitation of this study involves the nature of the control brain tissue used from autopsied infants (5). Virtually, all infants dying in the preterm age range have severe (lethal) disease processes that involve terminal hypoxia-ischemia and thus may not be truly representative of normal living infants. Despite the complexity in systemic disease processes and agonal conditions, there was no significant cerebral white matter pathology on standard histologic examination in any of our control cases.
The fate and ultimate function of the migrating DCX+ neurons in PVL are unknown. Our finding of increased DCX+ neurons in the white matter suggests that they are en route to compensate for the loss of white matter neurons, which in the human brain during late gestation are mainly subplate neurons or late migrating GABAergic neurons. Subplate neurons play a critical role in the establishment of connections between the cortex and thalamus, the development of ocular dominance column formation, and the cortical lamination (26–32). Although the subplate is a transient structure with major dissolution in the late fetal and early postnatal periods (30–32), a subset of its neurons persists in the adult white matter and is involved in the function of the mature cortex itself (27). In the human forebrain, GABAergic neurons continue to migrate to at least 27 wk (33) and possibly later (34), a hypothesis supported in this study by the presence of DCX+ cells in the white matter of our controls at term. Supporting the hypothesis that DCX+ cells are replacing GABAergic late migrating neurons in this study is the expression of GAD67/65, the immunomarker of the GABAergic phenotype, in a population of DCX+ cells. Colocalization of GABAergic markers (GABA and GAD) with DCX is seen in the cat, guinea pig, and primate cerebral cortex, as well as the human olfactory bulb (35–38), consistent with the differentiation of these immature cells into the GABAergic phenotype (35–38). In PVL, successful maturation of immature neurons, and incorporation of the DCX+ cells into the neuronal circuitry of the white matter, likely depends on the timing and extent of injury, and the cellular environment and availability of neurotrophic factors necessary for cellular differentiation. Innate repair mechanisms may not achieve complete repair of the neuronal damage. Thus, augmentation of this restorative attempt with growth factors and/or stem cells may be necessary to prevent the clinical neurological deficits in long-term survivors of PVL. This study yields evidence suggesting that innate repair is indeed underway in PVL to replenish the deficiency of white matter neurons needed to ensure proper development of the cerebral cortex.
Abbreviations
- DCX:
-
doublecortin
- EP:
-
encephalopathy of prematurity
- GFAP:
-
glial fibrillary acidic protein
- PVL:
-
periventricular leukomalacia
- SVZ:
-
subventricular zone
References
Volpe JJ 2009 Brain injury in premature infants: a complex amalgam of destructive and developmental disturbances. Lancet Neurol 8: 110–124
Petrini JR, Dias T, McCormick MC, Massolo ML, Green NS, Escobar GJ 2009 Increased risk of adverse neurological development for late preterm infants. J Pediatr 154: 169–176
Volpe JJ 2008 Neurology of the Newborn. WB Saunders Company, Philadelphia, PA
Kinney HC, Volpe JJ 2009 Perinatal panencephalopathy in the premature infants: is it due to hypoxia-ischemia? In: Haddad GG, Ping YS (eds) Brain Hypoxia and Ischemia: With Special Emphasis on Development. Humana Press, New York, NY, pp 153–186
Kinney HC 2009 The encephalopathy of prematurity: one pediatric neuropathologist's perspective. Semin Pediatr Neurol 16: 179–190
Back SA 2006 Perinatal white matter injury: the changing spectrum of pathology and emerging insights into pathogenetic mechanisms. Ment Retard Dev Disabil Res Rev 12: 129–140
Kadhim H, Khalifa M, Deltenre P, Casimir G, Sebire G 2006 Molecular mechanisms of cell death in periventricular leukomalacia. Neurology 67: 293–299
Jin K, Peel AL, Mao XO, Xie L, Cottrell BA, Henshall DC, Greenberg DA 2004 Increased hippocampal neurogenesis in Alzheimer's disease. Proc Natl Acad Sci U S A 101: 343–347
Chang A, Smith MC, Yin X, Fox RJ, Staugaitis SM, Trapp BD 2008 Neurogenesis in the chronic lesions of multiple sclerosis. Brain 131: 2366–2375
Yang Z, Levison SW 2007 Perinatal hypoxic/ischemic brain injury induces persistent production of striatal neurons from subventricular zone progenitors. Dev Neurosci 29: 331–340
Yang Z, Levison SW 2006 Hypoxia/ischemia expands the regenerative capacity of progenitors in the perinatal subventricular zone. Neuroscience 139: 555–564
Ong J, Plane JM, Parent JM, Silverstein FS 2005 Hypoxic-ischemic injury stimulates subventricular zone proliferation and neurogenesis in the neonatal rat. Pediatr Res 58: 600–606
Ikeda T, Iwai M, Hayashi T, Nagano I, Shogi M, Ikenoue T, Abe K 2005 Limited differentiation to neurons and astroglia from neural stem cells in the cortex and striatum after ischemia/hypoxia in the neonatal rat brain. Am J Obstet Gynecol 193: 849–856
Felling RJ, Snyder MJ, Romanko MJ, Rothstein RP, Ziegler AN, Yang Z, Givogri MI, Bongarzone ER, Levison SW 2006 Neural stem/progenitor cells participate in the regenerative response to perinatal hypoxia/ischemia. J Neurosci 26: 4359–4369
Plane JM, Liu R, Wang TW, Silverstein FS, Parent JM 2004 Neonatal hypoxic-ischemic injury increases forebrain subventricular zone neurogenesis in the mouse. Neurobiol Dis 16: 585–595
Okoshi Y, Itoh M, Takashima S 2001 Characteristic neuropathology and plasticity in periventricular leukomalacia. Pediatr Neurol 25: 221–226
Okoshi Y, Mizuguchi M, Itoh M, Oka A, Takashima S 2007 Altered nestin expression in the cerebrum with periventricular leukomalacia. Pediatr Neurol 36: 170–174
Tzarouchi LC, Astrakas LG, Zikou A, Xydis V, Kosta P, Andronikou S, Argyropoulou MI 2009 Periventricular leukomalacia in preterm children: assessment of grey and white matter and cerebrospinal fluid changes by MRI. Pediatr Radiol 39: 1327–1332
des Portes V, Pinard JM, Billuart P, Vinet MC, Koulakoff A, Carrie A, Gelot A, Dupuis E, Motte J, Berwald-Netter Y, Catala M, Kahn A, Beldjord C, Chelly J 1998 A novel CNS gene required for neuronal migration and involved in X-linked subcortical laminar heterotopia and lissencephaly syndrome. Cell 92: 51–61
Gleeson JG, Allen KM, Fox JW, Lamperti ED, Berkovic S, Scheffer I, Cooper EC, Dobyns WB, Minnerath SR, Ross ME, Walsh CA 1998 Doublecortin, a brain-specific gene mutated in human X-linked lissencephaly and double cortex syndrome, encodes a putative signaling protein. Cell 92: 63–72
Couillard-Despres S, Winner B, Schaubeck S, Aigner R, Vroemen M, Weidner N, Bogdahn U, Winkler J, Kuhn HG, Aigner L 2005 Doublecortin expression levels in adult brain reflect neurogenesis. Eur J Neurosci 21: 1–14
Gerber J, Tauber SC, Armbrecht I, Schmidt H, Bruck W, Nau R 2009 Increased neuronal proliferation in human bacterial meningitis. Neurology 73: 1026–1032
Kinney HC, Armstrong DL 2002 Perinatal neuropathology. In: Graham DI, Lantos PE (eds) Greenfield's Neuropathology. Arnold, London, United Kingdom, 557–559
Billiards SS, Haynes RL, Folkerth RD, Borenstein NS, Trachtenberg FL, Rowitch DH, Ligon KL, Volpe JJ, Kinney HC 2008 Myelin abnormalities without oligodendrocyte loss in periventricular leukomalacia. Brain Pathol 18: 153–163
González-Martínez JA, Bingaman WE, Toms SA, Najm IM 2007 Neurogenesis in the postnatal human epileptic brain. J Neurosurg 107: 628–635
Bystron I, Blakemore C, Rakic P 2008 Development of the human cerebral cortex: Boulder Committee revisited. Nat Rev Neurosci 9: 110–122
Clancy B, Silva-Filho M, Friedlander MJ 2001 Structure and projections of white matter neurons in the postnatal rat visual cortex. J Comp Neurol 434: 233–252
Chun JJ, Shatz CJ 1989 The earliest-generated neurons of the cat cerebral cortex: characterization by MAP2 and neurotransmitter immunohistochemistry during fetal life. J Neurosci 9: 1648–1667
Chun JJ, Shatz CJ 1989 Interstitial cells of the adult neocortical white matter are the remnant of the early generated subplate neuron population. J Comp Neurol 282: 555–569
Kostović I, Jovanov-Milosevic N 2008 Subplate zone of the human brain: historical perspective and new concepts. Coll Antropol 32: 3–8
Kostovic I, Rakic P 1980 Cytology and time of origin of interstitial neurons in the white matter in infant and adult human and monkey telencephalon. J Neurocytol 9: 219–242
Kostovic I, Rakic P 1990 Developmental history of the transient subplate zone in the visual and somatosensory cortex of the macaque monkey and human brain. J Comp Neurol 297: 441–470
Letinic K, Zoncu R, Rakic P 2002 Origin of GABAergic neurons in the human neocortex. Nature 417: 645–649
Robinson S, Li Q, Dechant A, Cohen ML 2006 Neonatal loss of gamma-aminobutyric acid pathway expression after human perinatal brain injury. J Neurosurg 104: 396–408
Cai Y, Xiong K, Chu Y, Luo DW, Luo XG, Yuan XY, Struble RG, Clough RW, Spencer DD, Williamson A, Kordower JH, Patrylo PR, Yan XX 2009 Doublecortin expression in adult cat and primate cerebral cortex relates to immature neurons that develop into GABAergic subgroups. Exp Neurol 216: 342–356
Xiong K, Luo DW, Patrylo PR, Luo XG, Struble RG, Clough RW, Yan XX 2008 Doublecortin-expressing cells are present in layer II across the adult guinea pig cerebral cortex: partial colocalization with mature interneuron markers. Exp Neurol 211: 271–282
Zhang XM, Cai Y, Chu Y, Chen EY, Feng JC, Luo XG, Xiong K, Struble RG, Clough RW, Patrylo PR, Kordower JH, Yan XX 2009 Doublecortin-expressing cells persist in the associative cerebral cortex and amygdala in aged nonhuman primates. Front Neuroanat 3: 17
Bédard A, Parent A 2004 Evidence of newly generated neurons in the human olfactory bulb. Brain Res Dev Brain Res 151: 159–168
Acknowledgements
We thank Mr. Richard A. Belliveau for assistance in this study.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Haynes, R., Xu, G., Folkerth, R. et al. Potential Neuronal Repair in Cerebral White Matter Injury in the Human Neonate. Pediatr Res 69, 62–67 (2011). https://doi.org/10.1203/PDR.0b013e3181ff3792
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/PDR.0b013e3181ff3792
This article is cited by
-
Mild intrauterine hypoperfusion reproduces neurodevelopmental disorders observed in prematurity
Scientific Reports (2016)
-
GABA receptors in brain development, function, and injury
Metabolic Brain Disease (2015)
-
A Diagnostic Approach for Cerebral Palsy in the Genomic Era
NeuroMolecular Medicine (2014)
-
Chronic lead exposure reduces doublecortin-expressing immature neurons in young adult guinea pig cerebral cortex
BMC Neuroscience (2012)
-
Diffuse periventricular leukomalacia in preterm children: assessment of grey matter changes by MRI
Pediatric Radiology (2011)
