
Abstract
Background
Phlebotomy-induced anemia (PIA) is common in premature infants and affects neurodevelopment. PIA alters hippocampal metabolism in neonatal mice through tissue hypoxia and iron deficiency. The mammalian target of rapamycin (mTOR) pathway senses the status of critical metabolites (e.g., oxygen, iron), thereby regulating hippocampal growth and function. We determined the effect of PIA and recombinant human erythropoietin (rHuEpo) treatment on mTOR signaling and expression of genes related to mTOR pathway functions.
Methods
Mice receiving an iron-supplemented diet were phlebotomized from postnatal day (P)3 to a target hematocrit of <25% by P7. Half were maintained at <25% until P14; half received rHuEpo from P7 to increase the hematocrit to 25–28%. Hippocampal phosphorylated to total protein ratios of four key mTOR pathway proteins were measured by western blotting at P14 and compared with non-phlebotomized, non-anemic control mice. mRNA levels of genes regulated by mTOR were measured by quantitative PCR.
Results
PIA suppressed phosphorylation of all mTOR proteins. rHuEpo restored AMP-activated protein kinase (AMPK) and AKT status, and partially rescued the mTOR output protein S6K. PIA and rHuEpo treatment also altered the expression of genes regulated by S6K.
Conclusion
PIA compromises and rHuEpo treatment partially rescues a pathway regulating neuronal DNA transcription, protein translation, and structural complexity.
Similar content being viewed by others
Main
Anemia occurs commonly and begins early in the neonatal time course in premature infants. It results in part from anemia of prematurity, but is greatly exacerbated by both necessary and unnecessary phlebotomy of these infants. Blood sampling for laboratory analysis can account for 10–40 ml/kg body weight per week; at its maximum, this is equivalent to removing the infant’s total blood volume every 2 weeks (1). Treatments for anemia include red blood cell transfusions and recombinant human erythropoietin (rHuEpo).
The risks and benefits of tolerating various degrees of anemia, as well as those of the two treatment strategies, have been well documented (1). Overall, the management of anemia in preterm infants remains empiric and controversial because the optimal and safest hemoglobin concentration is unknown. Although neonates can tolerate significant anemia from a cardiovascular stability standpoint, there is increasing evidence that the degree of anemia commonly tolerated in the NICU has the potential to negatively affect brain development. The concern stems from two randomized controlled trials that failed to clarify the relationships among the degree of anemia, treatments of anemia with red cell transfusions or rHuEpo, and neurodevelopmental outcomes. The multicenter Premature Infants in Need of Transfusion (PINT) trial showed a trend toward better short-term neurodevelopment in preterm infants randomized to receive treatment at a higher target hematocrit concentration (2), whereas the single-center Iowa trial showed better long-term outcomes in the infants previously randomized to a lower target (3). Because of the concern over transfusion-associated organ injuries (4, 5, 6), the overall trend in clinical practice has been to limit transfusions and tolerate lower hematocrit concentrations in spite of the potential risks of tissue hypoxia and iron deficiency to the developing brain. Preclinical modeling of phlebotomy-induced anemia (PIA) in lambs and mice indicate that the degree of anemia seen in preterm infants prioritizes iron to the red cells at the expense of the brain (7, 8, 9) and causes increased lactate and decreased pre-myelin fatty acid concentrations in the rapidly developing mouse hippocampus (8).
Mammalian target of rapamycin (mTOR) is a highly conserved protein kinase pathway that regulates important cell functions including cell size, survival, protein translation rates, and actin polymerization (Figure 1) (10, 11, 12). The pathway is highly active in developing hippocampal neurons and regulates dendrite arbor complexity, which in turn is linked to hippocampal function such as declarative learning and memory (13). mTOR signaling is driven largely by the availability of metabolic and structural substrates including oxygen, iron, branched-chain amino acids, and zinc (11, 12, 13). Disruption of mTOR signaling by neonatal iron deficiency anemia (IDA) in rodent models results in the suppression of mTOR signaling (14, 15) and dendrite structure abnormalities (16, 17). In contrast, non-anemic neonatal iron deficiency upregulates mTOR signaling, and also results in structural and behavioral abnormalities (13). The structural and behavioral abnormalities are reversed by normalization of pathway signaling through administration of rapamycin (13). Therefore, the balance of mTOR pathway activity in developing neurons is critical for normal neuronal differentiation and function to prevent long-term neurodevelopmental deficits.

The mammalian target of rapamycin pathway. A kinase-driven pathway regulating cell growth and proliferation through sensing of metabolic substrates such as oxygen, iron, and growth factors. Activity in the pathway was assessed through various phosphorylation sites on the proteins indicated (P). Boxed labels indicate an assessment in this study at either the mRNA or protein level.
The effects of PIA and its treatment with rHuEpo on neuronal mTOR signaling are not known. As PIA results in some of the same metabolic deficits as dietary IDA, we hypothesized that PIA itself would suppress mTOR activity and alter the expression of downstream target genes indexing its output and neuronal function. We expected that attenuation of PIA by rHuEpo treatment would increase the hematocrit, but could worsen brain iron deficiency, and therefore would not resolve all of the protein and gene expression abnormalities. The present study used target hematocrits similar to those in the PINT and Iowa trials in a neurodevelopmentally appropriate preclinical model of PIA to assess these effects on mTOR signaling and gene expression in the developing hippocampus.
Methods
Animal Preparation
The study was approved by the Institutional Animal Care and Use Committee at the University of Minnesota. Pregnant and lactating C57 B6 mice were fed and given water ad libitum, and maintained on a 14 h light/10 h dark cycle. Environmental temperature and humidity were controlled as per Research Animal Resources guidelines. An iron-supplemented rodent chow with 200 ppm of iron (Harlan; Indianapolis, IN) was fed throughout the experiment. This level of iron supplementation is 5 × the maintenance dose (18) and translates to a weight-based dose of 3–4 mg/kg body weight daily (19). Although this dose is similar to the standard iron dose for preterm infants, it is slightly lower than the dose used in trials of rHuEpo in neonates (20). Higher doses of iron have been shown to be toxic to the brain in rodents and were therefore not used (19, 21). Litters were culled to eight pups on P2 to reduce variability in food access and growth rate (8). A total of seven litters were used.
Induction of Anemia
Anemia was induced by phlebotomy via a micropipette through the facial vein as described previously (8, 22). Pups were weighed daily to determine the quantity of blood to be drawn with a goal minimum volume of 5.25 μl/g body weight. The hematocrit goal of <25% was based on levels used in the more transfusion-restricted groups in both the PINT and Iowa trials (23, 24), but also in consideration of normal neonatal mouse physiology. Both mice and humans are born relatively polycythemic and show a physiologic anemia of similar proportionality and hematocrit (8). The PIA groups required twice-daily phlebotomy until P7 and once-daily phlebotomy of 3.5 μl/g thereafter to maintain hematocrits within the target range. Hematocrits were measured daily by centrifugation of the microhematocrit collection tubes and quantification by hematocrit card reader. Non-phlebotomized control animals were handled similarly to phlebotomized animals and received a non-phlebotomizing needle stick daily.
Treatment with rHuEpo
The goal of rHuEpo treatment was to raise the hematocrit to a target of 25–28% to approximate the levels utilized in the more transfusion-liberal groups in the PINT and Iowa trials (23, 24). To achieve this goal, half of the PIA mice were treated with rHuEpo at a dose 2,000 IU/kg body weight twice daily beginning at P7 when the individual animal reached the target hematocrit of <25%. This dose was selected after initial experiments indicated that a previously published dose of 1,200 IU/kg in rats failed to significantly raise the hematocrit to the new target range of 25–28% (data not shown). Mice in both the PIA and PIA+rHuEpo groups continued to be phlebotomized once daily from P7 to P14 to maintain the target hematocrit concentrations and to realistically model conditions of continued phlebotomy losses in hospitalized premature infants.
Brain Collection and Processing
Mice were killed with an overdose of 120 mg/kg body weight of pentobarbital intraperitoneally on P14, the developmental equivalent of 40–44 weeks post-conceptional age in the human. The brain was removed and hippocampi were dissected and frozen in liquid nitrogen for storage at −80 °C before further processing.
Protein Analysis
Individual hippocampi were sonicated in lysis buffer (10 mM Tris, pH 7.4, 100 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM NaF, 20 mM Na 4 P 2 O 7, 2 mM Na 3 VO 4, 1% Triton X-100, 10% glycerol, 0.1% SDS, and 0.5% deoxycholate) with Complete protease inhibitor cocktail (Roche, Indianapolis, IN), as we have previously published (14). Reducing agent (Thermo Scientific, Waltham, MA) and LDS (Thermo) were added to each sample. Overall, 20–30 μg of total protein was separated using NuPAGE 4–12% Bis-Tris gels (Thermo) and transferred onto nitrocellulose membrane (Bio-Rad, Hercules, CA) as previously described (13). Using Rockland (Pottstown, PA) Blocking Buffer for Fluorescent Western Blotting and the following antibodies, the blots were imaged and analyzed with Odyssey infrared scanning (LiCor Bioscience, Lincoln, NE) as previously described. Mouse monoclonal anti-β-actin antibody was purchased from Sigma-Aldrich (St. Louis, MO) and used at a ratio of 1:10,000. Primary antibodies were purchased from Cell Signaling Technology (Danvers, MA) and used at a ratio of 1:500, which included pS6K (Thr389, #9205), S6K (#9202), pAkt (Ser473, #9271), pAkt (Thr308, #9275), Akt (#9272), p-mTOR (Ser2448, #2971), mTOR (#2972), pAMPKα (Thr172, #2531), and AMPKα (#2532). Secondary antibodies were Alexa Fluor 680-conjugated anti-mouse IgG (1:12,500, Thermo) and Infrared Dye 800-conjugated anti-rabbit IgG (1:12,500, Rockland).
Real-Time Quantitative PCR
RNA was extracted from the left hippocampus using the RNAqueous Total RNA Isolation Kit (Ambion, Austin, TX). The kit protocol was followed and samples were stored at −80 °C. Quality of samples was verified using a NanoDrop-2000 spectrophotometer (Thermo). Samples not meeting quality standards were further processed using the RNA Clean and Concentrator-5 Kit (Zymo Research, Irvine, CA). cDNA was generated using 1 μg of total RNA per sample and the High-Capacity RNA-to-cDNA kit (Invitrogen, Carlsbad, CA). Again the kit protocol was followed, and samples were diluted and stored at −20 °C. Quantitative PCR was run in duplex using Taqman qPCR Universal Master Mix and Taqman Gene Expression Assay probes (Applied Biosystems, Carlsbad, CA). Ribosomal protein s18 was used as an internal loading control. Thermocycling was performed with the MX3000P instrument (Strategene, La Jolla, CA).
Data analysis
A total of 11 non-phlebotomized control, 15 PIA, and 12 PIA+rHuEpo-treated mice across seven litters were used for western blot studies. To capture kinase activity within the mTOR pathway, western blot outcomes are presented as the ratio of phosphorylated protein to total protein for each protein assessed within the pathway. This was achieved by first using antibodies for the phosphorylated protein, and then using an antibody to the total protein on the same blot. Eight animals per group were used in the mRNA experiments. mRNA expression of the non-phlebotomized controls was set at 1, and the expression in the experimental groups was recorded as a ratio of experimental to control. Comparisons among groups were assessed by one-way ANOVA with post-hoc t-tests. Differences were considered significant at P<0.05.
Results
The hematocrits at P14 of the PIA group were 34% lower than the nonbled control group. The group receiving rHuEpo treatment (PIA+rHuEpo) was significantly different from both the PIA group and the non-phlebotomized control group with an average hematocrit 21% lower than the nonbled group (Table 1). Weight gain was not statistically different across all groups (Table 1).
PIA Induced Hippocampal Iron Deficiency and Tissue Hypoxia
P14 hippocampal tissues were analyzed for changes in the expression of genes known to respond to iron and/or oxygen status. Iron status was assessed through expression of transferrin receptor 1 (Tfrc1). Tfrc1 was increased by 50% in the PIA group (Figure 2a), a level similar to that seen in dietary models of iron deficiency (14). Tfrc1 expression remained elevated in the PIA+rHuEpo group, indicating ongoing hippocampal iron deficiency (Figure 2a). Hippocampal expression of vascular endothelial growth factor (VEGF), which responds to changes in oxygen status via Hif1a, was increased in the PIA group (Figure 2b), indicating that a hematocrit of <25% induces a significant degree of hippocampal hypoxia. rHuEpo treatment partially, but not completely, rescued VEGF expression (Figure 2b). VEGF expression between the PIA and PIA+rHuEpo group was not statistically different.

Phlebotomy-induced anemia (PIA) induces iron deficiency and tissue hypoxia in P14 mouse hippocampus. (a) Transferrin receptor (Tfrc) expression was increased in PIA mice (light gray) as compared with nonbled control mice (black), an indication of iron deficiency. Treatment with rHuEpo (dark gray) resulted in no change in transferrin receptor mRNA expression. (b) VEGF expression was increased in PIA mice as compared with nonbled control mice, indicating tissue hypoxia. Treatment with rHuEpo resulted in a partial return toward nonbled control levels. Values are mean±SEM normalized to the control group. n=8 per group. *P<0.05, †P<0.001 for each comparison. rHuEpo, recombinant human erythropoietin; VEGF, vascular endothelial growth factor.
PIA Suppressed mTOR Protein Phosphorylation in the Developing Hippocampus
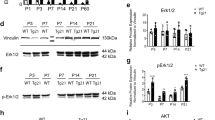
In the mTOR pathway, two branches provide critical input for mTOR regulation (Figure 1). The first input branch is a growth factor regulated input pathway, in which PI3K signaling mediates phosphorylation of AKT. In our model, the phosphorylated AKT to total AKT ratio was significantly decreased in the PIA group (Figure 3a). rHuEpo treatment restored this ratio to non-phlebotomized control levels (Figure 3a). The second input branch of the pathway integrates signals related to energy status and oxygen availability through phosphorylation of AMP-activated protein kinase (AMPK). PIA resulted in a significant decrease in the ratio of phosphorylated AMPK to total AMPK, which was similarly fully rescued by rHuEpo treatment (Figure 3b).

mTOR pathway protein expression. (a) Expression of pAKT/AKT was decreased in PIA mice (light gray) as compared with nonbled control mice (black). Treatment with rHuEpo resulted in ratios similar to nonbled control levels. (b) pAMPK/AMPK was decreased in PIA mice and fully recovered with rHuEpo treatment. (c) pMTORC1/MTORC1 expression is decreased in PIA mice and remains decreased with rHuEpo treatment as compared with nonbled control mice. (d) pS6K/S6K expression is decreased in PIA mice and partially recovered by rHuEpo treatment. Values are mean±SEM normalized to the control group. n=9–15 per group. *P<0.05, †P<0.001 for each comparison. PIA, phlebotomy-induced anemia; rHuEpo, recombinant human erythropoietin; AMPK, AMP-activated protein kinase; MTORC1, mammalian target of rapamycin complex 1.
mTOR complex 1 is a critical integration component of the mTOR signaling pathway (Figure 1). The ratio of the phosphorylated mTORC1 to total mTORC1 protein was significantly decreased in the PIA group, but was not rescued by rHuEpo treatment (Figure 3c).
The effector portion of the mTOR pathway includes the protein kinase S6K, the activity of which determines the net output of the pathway. PIA significantly suppressed S6K activity compared with the nonbled controls (Figure 3d). rHuEpo treatment significantly increased the ratio of phosphorylated to total S6K (Figure 3d).
Treatment of PIA with rHuEpo Increased the Expression of Genes Regulated by S6K
Output from the mTOR pathway was assessed through changes in the expression of several genes indexing functions regulated by S6K, including cell cycle progression, neuronal protein translation, and DNA transcription. Cyclin D1 (Ccnd1) and cyclin-dependent kinase 4 (Cdk4) are directly involved in the regulation of cell cycle progression leading to increased cell growth. Both Ccnd1 and Cdk4 expression levels were increased in the PIA+rHuEpo-treated animals as compared with the PIA group (Figure 4a). Eukaryotic translation initiation factor 4b (Eif4b) is required for the binding of mRNA to ribosomes leading to an increase in protein synthesis. Eif4b was significantly higher in the PIA+rHuEpo group as compared with PIA alone (Figure 4c). Finally, cAMP responsive element-binding protein 1 (Creb1), an important transcriptional regulator, was also increased in the PIA+rHuEpo-treated group as compared with the PIA only group (Figure 4d).

Genes regulated by mTOR output protein S6K. (a) Cyclin D1 (Ccnd1) expression is increased in hippocampus of P14 mice treated with rHuEpo. (b) Cyclin-dependent kinase 4 (Cdk4) expression is increased in rHuEpo-treated hippocampus. (c,d) Expression of eukaryotic translation initiation factor 4b (Eif4b, c) and cAMP responsive element-binding protein 1 (Creb1, d) are increased in the PIA+rHuEpo group as compared with PIA alone. Values are mean±SEM normalized to the nonbled control group. n=6–8 per group. *P<0.05, **P<0.01 for each comparison. PIA, phlebotomy-induced anemia; rHuEpo, recombinant human erythropoietin.
Discussion
Premature birth remains a major cause of neurobehavioral disabilities in childhood. Although many factors influence neurodevelopment in preterm infants, relatively few are within the control of the neonatologist. However, two highly controllable factors that can affect neurodevelopment are the degree of anemia (3, 23, 24, 25, 26) and the treatment strategies to relieve anemia such as rHuEpo. Neonatal phlebotomy remains the major cause of anemia in preterm infants in the NICU (1).
Randomized trials that assess neurodevelopment as a function of neonatal anemia are relatively scarce and their results are equivocal. In a single-center trial based at the University of Iowa and in the multicenter PINT trial in Canada, short-term neurodevelopmental outcomes were more favorable in infants randomized to receive treatment at a higher hematocrit threshold (2). However, long-term follow-up of the Iowa trial cohort revealed better outcomes in the group with a lower hematocrit threshold for treatment (3). Thus, the optimal hematocrit for preterm neonates remains unclear.
In the current study, we used a previously validated physiologically relevant mouse model of PIA to assess its effects on the developing brain (8). The hematocrit levels of the two experimental groups were designed to closely approximate the treatment thresholds used in the human trials (23, 24). In addition, mice and humans have similar hematocrits at birth, are both relatively polycythemic at birth, and both undergo a process of physiologic anemia (8). The onset and duration of anemia in the mouse were timed to be typical for a preterm newborn in the NICU, with P3 in the mouse approximating a 26-week post-conceptional age brain in the human and P14 approximating term. This time period coincides with a period of rapid hippocampal development in both preterm humans and neonatal mice (27). The increase of VEGF in the PIA hippocampus is indicative of tissue-level hypoxia because of the induced anemia. We have previously measured arterial blood gases in this model and found no differences in blood gases (data not shown) or evidence of lactic acidosis between PIA and nonbled mice, indicating that hypoxia and lactate effects are seen in the brain before the periphery. Anemia significantly affects hippocampal oxygenation and iron status (8, 14). Both oxygen and iron are critical substrates for energy metabolism in the developing brain, which accounts for close to 60% of the total body oxygen consumption in the neonate (28). The complexity of the brain’s structural development, which confers its functional capacity, is highly dependent on adequate oxygen and iron availability. These substrates regulate brain structural development through signaling pathways such as mTOR (10, 12, 13).
The mTOR pathway is a tightly regulated kinase signaling pathway that integrates multiple metabolic inputs including oxygen, iron, glucose, zinc, and amino acids to control neuronal growth and survival through regulation of protein translation, DNA transcription, autophagy, and actin polymerization (10, 12, 13). Optimal mTOR activity is required for normal neurodevelopment with either low or excessive mTOR activity resulting in abnormal dendrite structure and function (29, 30).
In the current study, a hematocrit of <25% reduced the activity of mTOR proteins in the upper (input), middle (integration), and lower (output) parts of the pathway in the mouse hippocampus. The effect may be driven by tissue hypoxia, iron deficiency (8), or both. The 50% increase in Tfrc1 gene expression compared with the non-anemic controls is consistent with an ~40% reduction in hippocampal iron concentration that has also been documented in a model of dietary IDA in neonatal rats (14) and in this model of PIA in neonatal mice (8). In the dietary IDA models, this degree of hippocampal iron deficiency causes significant reduction in mTOR pathway activity (14) accompanied by both structural and functional dendritic abnormalities (16, 31, 32, 33). This degree of hippocampal iron deficiency occurred in the PIA model in spite of an iron-supplemented diet that provided four times the recommended amount of iron (18) with an estimated dose of 3–4 mg/kg of elemental iron to the pups daily (19).
Treatment of PIA with rHuEpo increased the hematocrit to 25–28%, thus partially relieving tissue hypoxia. rHuEpo normalized the upper elements of the mTOR pathway to control, nonbled values, but the middle and lower elements remained partially suppressed. Contrary to our expectation that available iron would be shunted into red cells at the expense of the brain (9, 34), the degree of hippocampal iron deficiency did not worsen. Nevertheless, evidence of significant brain iron deficiency remained present in the rHuEpo-treated group with continued elevation of Tfrc1 expression. These findings suggest that the upper elements of the pathway are driven more by tissue hypoxia, whereas the integration portion is driven more by iron deficiency. As the output element (S6K) was partially rescued, it may be driven by both tissue hypoxia and iron deficiency.
Our previous investigation in this model using proton nuclear magnetic resonance spectroscopy in the living animal demonstrated that a PIA-induced hematocrit of <25% alters hippocampal energy metabolism (8). The elevated lactate concentrations and reduced phosphocreatine to creatine ratios found in that study indicate compromised tricarboxylic acid cycle (TCA) activity and mitochondrial oxidative phosphorylation. Multiple enzymes in the TCA cycle are iron dependent, whereas oxidative phosphorylation is both iron and oxygen dependent. We propose that compromise of both biochemical processes likely led to the observed reduction in the ratio of phosphorylated AMPK to total AMPK protein in the current study. We further speculate that tissue hypoxia was the main driving factor of the AMPK alterations, as correction of anemia but not brain iron deficiency fully restored AMPK activity.
Similar to AMPK, HIF1a stimulates TSC 1/2, which resides in the integrative part of the mTOR pathway (Figure 1) (10, 12). HIF1a is regulated by oxygen and iron status, together or independently, and increases VEGF mRNA under conditions of either hypoxia or iron deficiency (31, 35). As VEGF gene expression was elevated in both the <25% hematocrit group and the less anemic rHuEpo-treated group, we speculate that this input into the integrative part of the mTOR pathway was controlled primarily by the tissue iron deficiency induced by PIA (8).
The third input into the integrative portion of the mTOR pathway consists of growth factors that regulate AKT phosphorylation (10, 11, 12). These include IGF-1 and brain-derived neurotrophic factor (BDNF). Both genes are downregulated in models of IDA, which are characterized by tissue hypoxia and brain iron deficiency (36, 37). In the current study, PIA reduced the ratio of phosphorylated AKT to total AKT protein, and was rescued by rHuEpo treatment. The findings are more consistent with hypoxia, rather than iron deficiency being the dominant driver of this effect. Collectively, the sum effects of PIA through these three inputs resulted in reduced activity of mTORC1, a key intermediary integrative step in the pathway that directly regulates the status of the output protein, S6K.
Both mTORC1 and S6K were suppressed by PIA, and S6K was only partially rescued by rHuEpo treatment. The failure of rHuEpo to completely reverse the suppression of downstream mTOR pathway elements could be because of only partial correction of the hematocrit, the persistence of tissue iron deficiency, or the short duration of treatment. S6K activity directly regulates protein translation rates, DNA transcription rates, and cell cycle progression. The upregulation of genes regulated by S6K that index these processes include Eif4b, Creb1, Ccnd1, and Cdk4. The observed upregulation of these genes by rHuEpo treatment may be an acute response to relief of tissue hypoxia through correction of anemia or through rHuEPo acting as a growth factor to stimulate the upper mTOR pathway elements via activation of PI3K signaling (38, 39). The compensatory increase in these genes appears to occur before the complete reversal of mTORC1 or S6K protein expression.
Collectively, the present study demonstrates that PIA to the degree frequently tolerated in preterm neonates presents a fundamental risk to metabolic processes regulating mTOR signaling in the developing mouse hippocampus that ultimately determines the area’s structural and functional capacity. Treatment of anemia with rHuEpo to achieve hematocrits that are closer to the liberal thresholds used in human studies of neonatal anemia (23, 24) spares some, but not all, of the metabolic processes. One potential translational limitation of this model is the difference in growth rate and cerebral metabolism of the mouse as compared with the human neonate. As the growth rate of the mouse is much faster than humans, this in turn puts more pressure on the iron balance as most iron is used for expansion of the red cell mass with growth. For this reason, the level of brain iron deficiency may be worse in mice than in humans. We also know that the relative metabolic rate of the brain (i.e., percentage of cerebral oxygen consumption as a function of total body oxygen consumption) is greater in humans than in mice. One of iron’s main roles in the brain is to support oxidative phosphorylation through its incorporation into cytochromes. This would indicate that the effects of anemia and brain iron deficiency in the human neonate may be greater than those we have seen in our study. Therefore, extrapolation of our data obtained at a hematocrit of <25% in mice to humans needs to take this balancing of red cell iron demand and brain iron demand into consideration. It is possible that targeting a higher hematocrit (e.g., 35%) or that increasing iron supplementation to address the ongoing iron deficiency in the rHuEpo-treated group could result in full recovery of the mTOR signaling pathway. An exciting next step will be to determine whether the protective neurodevelopmental effects of erythropoietin that have been shown in human studies (20, 40) are also present in our model.
References
Widness JA . Pathophysiology of anemia during the neonatal period, including anemia of prematurity. NeoReviews 2008;9:e520.
Whyte RK, Kirpalani H, Asztalos EV et al, Neurodevelopmental outcome of extremely low birth weight infants randomly assigned to restrictive or liberal hemoglobin thresholds for blood transfusion. Pediatrics 2009;123:207–13.
McCoy TE, Conrad AL, Richman LC, Lindgren SD, Nopoulos PC, Bell EF . Neurocognitive profiles of preterm infants randomly assigned to lower or higher hematocrit thresholds for transfusion. Child Neuropsychol 2011;17:347–67.
Kelly AM, Williamson LM . Neonatal transfusion. Early Hum Dev 2013;89:855–60.
La Gamma EF, Blau J . Transfusion-related acute gut injury: feeding, flora, flow, and barrier defense. Semin Perinatol 2012;36:294–305.
Singh R, Shah BL, Frantz ID . Necrotizing enterocolitis and the role of anemia of prematurity. Semin Perinatol 2012;36:277–82.
Guiang SF 3rd, Georgieff MK, Lambert DJ, Schmidt RL, Widness JA . Intravenous iron supplementation effect on tissue iron and hemoproteins in chronically phlebotomized lambs. Am J Physiol 1997;273:R2124–31.
Wallin DJ, Tkac I, Stucker S et al, Phlebotomy-induced anemia alters hippocampal neurochemistry in neonatal mice. Pediatr Res 2015;77:765–71.
Zamora TG, Guiang SF, Widness JA, Georgieff MK . Iron is prioritized to red blood cells over the brain in phlebotomized anemic newborn lambs. Pediatr Res 2016;79:922–8.
Fretham SJB, Carlson ES, Georgieff MK . The role of iron in learning and memory. Adv Nutr 2011;2:112–21.
Meijer AJ, Lorin S, Blommaart EF, Codogno P . Regulation of autophagy by amino acids and MTOR-dependent signal transduction. Amino Acids 2015;47:2037–63.
Wullschleger S, Loewith R, Hall MN . TOR signaling in growth and metabolism. Cell 2006;124:471–84.
Fretham SJB, Carlson ES, Georgieff MK . Neuronal-specific iron deficiency dysregulates mammalian target of rapamycin signaling during hippocampal development in nonanemic genetic mouse models. J Nutr 2013;143:260–6.
Carlson ES, Stead JDH, Neal CR, Petryk A, Georgieff MK . Perinatal iron deficiency results in altered developmental expression of genes mediating energy metabolism and neuronal morphogenesis in hippocampus. Hippocampus 2007;17:679–91.
Ndong M, Kazami M, Suzuki T et al, Iron deficiency down-regulates the Akt/TSC1-TSC2/mammalian target of rapamycin signaling pathway in rats and in COS-1 cells. Nutr Res 2009;29:640–7.
Brunette KE, Tran PV, Wobken JD, Carlson ES, Georgieff MK . Gestational and neonatal iron deficiency alters apical dendrite structure of CA1 pyramidal neurons in adult rat hippocampus. Dev Neurosci 2010;32:238–48.
Jorgenson LA, Wobken JD, Georgieff MK . Perinatal iron deficiency alters apical dendritic growth in hippocampal CA1 pyramidal neurons. Dev Neurosci 2003;25:412–20.
Felt BT, Beard JL, Schallert T et al, Persistent neurochemical and behavioral abnormalities in adulthood despite early iron supplementation for perinatal iron deficiency anemia in rats. Behav Brain Res 2006;171:261–70.
Unger EL, Hurst AR, Georgieff MK et al, Behavior and monoamine deficits in prenatal and perinatal iron deficiency are not corrected by early postnatal moderate-iron or high-iron diets in rats. J Nutr 2012;142:2040–9.
Ohls RK, Cannon DC, Phillips J et al, Preschool assessment of preterm infants treated with darbepoetin and erythropoietin. Pediatrics 2016;137:e20153859.
Rao R, Tkac I, Unger EL et al, Iron supplementation dose for perinatal iron deficiency differentially alters the neurochemistry of the frontal cortex and hippocampus in adult rats. Pediatr Res 2013;73:31–7.
Liu Z-J, Hoffmeister KM, Hu Z et al, Expansion of the neonatal platelet mass is achieved via an extension of platelet lifespan. Blood 2014;123:3381–9.
Bell EF, Strauss RG, Widness JA et al, Randomized trial of liberal versus restrictive guidelines for red blood cell transfusion in preterm infants. Pediatrics 2005;115:1685–91.
Kirpalani H, Whyte RK, Andersen C et al, The premature infants in need of transfusion (PINT) study: a randomized, controlled trial of a restrictive (low) versus liberal (high) transfusion threshold for extremely low birth weight infants. J Pediatr 2006;149:301–7.
McCoy TE, Conrad AL, Richman LC et al, The relationship between brain structure and cognition in transfused preterm children at school age. Dev Neuropsychol 2014;39:226–32.
Nopoulos PC, Conrad AL, Bell EF et al, Long-term outcome of brain structure in premature infants: effects of liberal vs restricted red blood cell transfusions. Arch Pediatr Adolesc Med 2011;165:443–50.
Rice D, Barone S Jr . Critical periods of vulnerability for the developing nervous system: evidence from humans and animal models. Environ Health Perspect 2000;108:511–33.
Kuzawa CW . Adipose tissue in human infancy and childhood: an evolutionary perspective. Am J Phys Anthropol 1998;27:177–209.
Ehninger D, Han S, Shilyansky C et al, Reversal of learning deficits in a Tsc2+/− mouse model of tuberous sclerosis. Nat Med 2008;14:843–848.
Zhou J, Blundell J, Ogawa S et al, Pharmacological inhibition of mTORC1 suppresses anatomical, cellular, and behavioral abnormalities in neural-specific Pten knock-out mice. J Neurosci 2009;29:1773–83.
Carlson ES, Tkac I, Magid R et al, Iron is essential for neuron development and memory function in mouse hippocampus. J Nutr 2009;139:672–9.
Jorgenson LA, Sun M, O’Connor M, Georgieff MK . Fetal iron deficiency disrupts the maturation of synaptic function and efficacy in area CA1 of the developing rat hippocampus. Hippocampus 2005;15:1094–102.
Schmidt AT, Waldow KJ, Grove WM, Salinas JA, Georgieff MK . Dissociating the long-term effects of fetal/neonatal iron deficiency on three types of learning in the rat. Behav Neurosci 2007;121:475–82.
Georgieff MK, Schmidt RL, Mills MM, Radmer WJ, Widness JA . Fetal iron and cytochrome c status after intrauterine hypoxemia and erythropoietin administration. Am J Physiol 1992;262:R485–91.
Siddiq A, Aminova LR, Ratan RR . Prolyl 4-hydroxylase activity-responsive transcription factors: from hydroxylation to gene expression and neuroprotection. Front Biosci J Virtual Libr 2008;13:2875–87.
Tran PV, Fretham SJB, Carlson ES, Georgieff MK . Long-term reduction of hippocampal brain-derived neurotrophic factor activity after fetal-neonatal iron deficiency in adult rats. Pediatr Res 2009;65:493–8.
Tran PV, Fretham SJB, Wobken J, Miller BS, Georgieff MK . Gestational-neonatal iron deficiency suppresses and iron treatment reactivates IGF signaling in developing rat hippocampus. Am J Physiol Endocrinol Metab 2012;302:E316–24.
Kretz A, Happold CJ, Marticke JK, Isenmann S . Erythropoietin promotes regeneration of adult CNS neurons via Jak2/Stat3 and PI3K/AKT pathway activation. Mol Cell Neurosci 2005;29:569–79.
Ma R, Xiong N, Huang C et al, Erythropoietin protects PC12 cells from β-amyloid25–35-induced apoptosis via PI3K/Akt signaling pathway. Neuropharmacology 2009;56:1027–34.
Bierer R, Peceny MC, Hartenberger CH, Ohls RK . Erythropoietin concentrations and neurodevelopmental outcome in preterm infants. Pediatrics 2006;118:e635–40.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Statement of financial support
This research was funded by the NIH Grant P01 HL046925 and NIH Grant R01 HD029421
Rights and permissions
About this article

Cite this article
Wallin, D., Zamora, T., Alexander, M. et al. Neonatal mouse hippocampus: phlebotomy-induced anemia diminishes and treatment with erythropoietin partially rescues mammalian target of rapamycin signaling. Pediatr Res 82, 501–508 (2017). https://doi.org/10.1038/pr.2017.88
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/pr.2017.88
This article is cited by
-
Dose- and sex-dependent effects of phlebotomy-induced anemia on the neonatal mouse hippocampal transcriptome
Pediatric Research (2022)
-
Transfusion prevention using erythropoietin, parenteral sucrose iron, and fewer phlebotomies in infants born at ≤30 weeks gestation at a high altitude center: a 10-year experience
Journal of Perinatology (2021)
-
Transfusions and neurodevelopmental outcomes in extremely low gestation neonates enrolled in the PENUT Trial: a randomized clinical trial
Pediatric Research (2021)
