
Abstract
Epidermolysis bullosa is classified as a genodermatosis, an inherited genetic skin disorder that results in severe, chronic skin blistering with painful and life-threatening complications. Although there is currently no cure for epidermolysis bullosa, concurrent advances in gene and stem cell therapies are converging toward combinatorial therapies that hold the promise of clinically meaningful and lifelong improvement. Recent studies using hematopoietic stem cells and mesenchymal stromal/stem cells to treat epidermolysis bullosa have demonstrated the potential for sustained, effective management of the most severe cases. Furthermore, advances in the use of gene therapy and gene-editing techniques, coupled with the development of induced pluripotent stem cells from patients with epidermolysis bullosa, allow for autologous therapies derived from a renewable population of cells that are patient-specific. Here we describe emerging treatments for epidermolysis bullosa and other genodermatoses, along with a discussion of their benefits and limitations as effective therapies.
Similar content being viewed by others


Main
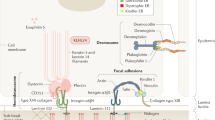
Epidermolysis bullosa (EB) belongs to a group of rare genetic skin disorders primarily caused by mutations in genes that encode for extracellular matrix proteins. EB is characterized by persistent skin blistering and painful lesions. Recessive dystrophic epidermolysis bullosa (RDEB), one of the most severe forms of EB, is due to loss-of-function mutations in the essential extracellular matrix protein type VII collagen (C7) (1). Loss or diminished function of C7 leads to weakness in the structural architecture of the dermal–epidermal junction (DEJ) and mucosal membranes where C7 is deposited (Figure 1). This loss of structural support leads to skin blistering and complications, including esophageal strictures, mitten deformities, itching, and painful blistering (2, 3). Children with RDEB are affected from birth, with little to no respite despite constant care and extensive wound dressing (4).

An illustration of the differences between healthy skin (a) and RDEB skin (b) at the dermal–epidermal junction (80). Copyright © 2015 Massachusetts Medical Society. Reprinted with permission. RDEB, recessive dystrophic epidermolysis bullosa.
In normal skin, fibroblasts and keratinocytes produce C7 near the DEJ and in response to injury or tissue damage (5, 6, 7). Through local coordination between systemic immune cells that regulate inflammatory and wound-healing responses, human skin is capable of regenerating injured tissue and maintaining a homeostatic state. However, in the case of RDEB, the supporting cells near the DEJ are unable to produce functional C7 and generate a normal healing response. Furthermore, chronic wounds such as those found in patients with RDEB have shown a limited ability to remodel the extracellular matrix in a productive manner, further limiting the ability for RDEB skin to regenerate itself.
Although many of the signs and symptoms of RDEB are apparent at birth, certain aspects of the disease pathology are progressive in nature. Loss of the physical barrier and disruption of the immunologic function of the skin leads to persistent chronic infections, acquired resistance to antibiotics, and can become refractory to interventional therapies. Sustained TGF-β activity and resulting contractile fibrosis from chronic wound healing leads to pseudosyndactyly (8). In addition, RDEB patients struggle with eating solid food, which leads to malnutrition, and develop corneal abrasions that worsen with time (9). Furthermore, many RDEB patients develop an aggressive form of squamous cell carcinoma later in life (10, 11, 12, 13). Due to the life-altering, severe nature of this systemic disease, stem cell therapies—with the potential to address the underlying cause of the disorder by providing a lifelong source of normal C7—should be considered.
Stem cell therapy for rdeb
Stem cells have the capacity to address the diverse nature of RDEB symptoms. Initial research in animal models showed that hematopoietic cell transplantation (HCT) of donor-derived cells contributed to wound healing in the skin (14). In this study, green fluorescent protein-labeled bone marrow was transplanted into non-green fluorescent protein-labeled mice following cutaneous wounding. Interestingly, wounding stimulated bone marrow cell engraftment and induced production of non-hematopoietic skin structures from bone marrow-derived cells. HCT in an RDEB mouse model demonstrated an improvement in animal survival and skin strength (15), and transplanted CD150+/48− cells homed to injured skin. Deposits of C7 protein and rudimentary anchoring fibrils—structures required for skin integrity that are absent in RDEB mice—were found in treated mice, providing evidence that a subset of cells of hematopoietic origin are capable of correcting the basement membrane zone defect in a murine RDEB model. This work was the foundational platform upon which the first-in-human studies were based (16). Six children with RDEB received allogeneic HCT, all of whom exhibited improved wound healing and a reduction in blister formation. Between 30 and 130 days after transplantation, an increase in C7 deposition was found at the DEJ in five of the six recipients; however, there was no observed normalization of the anchoring fibrils at the times measured. In all six recipients, substantial numbers of donor cells were found in the skin. This initial experience not only supported the potential effectiveness of allogeneic HCT but also the future use of gene-corrected autologous hematopoietic stem cells (17). Although HCT appears to be a viable option for RDEB patients, the inherent risks and safety concerns of HCT are still present, and other approaches with stem cells of non-hematopoietic origin could be used as an alternative or auxiliary approach to HCT. Because of the role of mesenchymal stromal/stem cells (MSCs) in wound healing and their beneficial effect in animal models of EB, we and others hypothesized that MSCs may play a supportive role in the transplant setting. Initial work studied the role of MSCs in wound healing (18). The differentiation potential of MSCs allowed them to be recruited to sites of skin injuries and to transdifferentiate into multiple skin cell types, improving the natural healing process. Preconditioning of murine MSCs increased expression of Col7a1 and demonstrated their utility for future transplant studies (19). Systemic application of bone marrow-derived MSCs in a 10-patient clinical trial showed moderated blister severity and surface area for 4–6 months after transfusion and an improved quality of life for the patients (20). Although HCT reduces the number and severity of blistering episodes, it is not a panacea. The mechanisms involved in these outcomes are poorly characterized. Some insight has been gained with the discovery of PDGFRα+ cells from bone marrow contributing to epithelial regeneration after skin grafting in mice (21). PDGFRα+ cells have also been found to restore C7 deposition in EB mouse models (22). Beyond identifying the inherent mechanism of how HCT is capable of improving RDEB, there are other questions regarding the long-term outcomes of HCT in these patients and whether changes in the HCT protocol can be made to reduce the severity of the conditioning regimen.
One of the major challenges facing the use of stem cells as a treatment option for RDEB is the donor source. In addition to expansion of skin cells (23, 24), the derivation of induced pluripotent stem cells (iPSCs) (25, 26) has opened the possibility of autologous cell transplantation in EB. This method has been demonstrated as a potential therapy in models of other diseases including sickle cell anemia, limb-girdle muscular dystrophy, and macular degeneration (27, 28, 29). Terminally differentiated skin and bone marrow cells from iPSCs appear to elicit little or no immune response in strain-matched murine models following transplant (30). Importantly, iPSCs have been derived from patients with RDEB (31) and junctional epidermolysis bullosa (32). This suggests an opportunity for personalized, patient-specific cell transfer. It also demonstrates that functional C7 is dispensable for stem cell renewal and differentiation into both hematopoietic and non-hematopoietic lineages. More recently, iPSCs have been derived from RDEB patient T cells (33). Although generating iPSCs from EB patients is an important first step, these cells still harbor the mutations in the COL7A1 gene. A promising option has been to reprogram naturally revertant skin cells (in which the genetic mutations were self-corrected) from RDEB patients (34, 35). For those without detectable gene reversion, gene-editing technology has emerged as a powerful corrective tool through zinc-finger nucleases (36). The continued advances in gene editing using TALEN and CRISPR/Cas9 have improved the speed and efficiency of deriving and correcting RDEB iPSCs (37, 38, 39).
The true potential of iPSCs to treat RDEB comes in deriving transplantable cells with functional C7. In addition, the ability of iPSCs to derive cells of the endoderm, mesoderm, and ectoderm makes them useful for RDEB and other diseases with widespread effects (40, 41). Generating functional hematopoietic cells from iPSCs that are capable of engraftment is also an area of research progress (42, 43, 44, 45). Local injection of fibroblasts into the margins of chronic erosions in RDEB patients increases wound healing for the first 28 days (46). However, after the initial improvement window, changes in healing are not significantly different between sites of fibroblast or vehicle injection.
As skin blistering and the resulting chronic open wounds are a major complication in RDEB patients, much work has been done to address this issue, from the formation of a three-dimensional skin equivalent tissue (47) to generation of keratinocytes from corrected RDEB iPSCs (39). Though promising, many hurdles exist in the use of iPSC-derived cells for autologous cell therapy, including genetic and epigenetic instability, and post-transplantation efficacy and safety (48).
Gene therapy for rdeb
The most common form of gene therapy for treating genetic disorders uses viral vectors. Retroviral, lentiviral, and adenoviral vectors have been developed for RDEB gene therapy. Retroviral vectors were used to transduce fibroblasts, which were then evaluated and used for injection into a mouse model of RDEB. Transduced fibroblasts were shown to express functional C7, deposit it as mature anchoring fibrils, and provide improvement based on both in vitro and in vivo evaluations (49). In similar studies, skin equivalents were also made from genetically corrected RDEB cells (50, 51). The first use of gene therapy for RDEB patients was a retroviral vector used to transduce keratinocytes containing full-length human COL7A1 (52). The transduced keratinocytes were then grown in a GMP facility to generate corrected epidermal sheets used for autologous therapy. These external autologous grafts were tolerated for 12 months with positive outcomes. Adenoviral vectors were similarly used to correct RDEB cells, both fibroblasts and keratinocytes, and subsequently to establish iPSC lines for future therapeutic use (38). Corrected iPSCs were then used to differentiate into keratinocytes that were capable of expressing C7 and developing into stratified layers both in vitro and in vivo. Lentiviral vectors have also been developed for C7 gene therapy (53). Recently, a lentiviral vector was developed containing a codon-optimized COL7A1 gene and was used to correct RDEB fibroblasts (54). Corrected fibroblasts were shown to express full-length, functional C7 in vitro and to deposit C7 at the DEJ in skin grafts on immunodeficient mice. These approaches have the potential to be useful in developing the combinatorial therapies needed to address the systemic problems of this disease.
Non-viral strategies have also been developed for correcting RDEB cells, including the use of synthetic knot polymers for genetic transfection (55). Synthetic knot polymers allow for efficient transfection and lower toxicity in comparison to standard transfection reagents. Transfection of RDEB keratinocytes led to efficient expression of C7 in vitro. The use of non-viral-mediated gene therapy also includes the use of transposons, including Sleeping Beauty, to correct RDEB cells. Full-length COL7A1 was recently inserted into RDEB keratinocytes via Sleeping Beauty transposons with stable integration (56). RDEB cells containing this cassette were capable of producing functional C7 in vitro and in vivo. Another non-viral approach for treating RDEB involves correcting parts of the mutated COL7A1 transcript through targeted exon skipping or trans-splicing. Exon skipping allows for truncated, functional C7 to be produced in RDEB cells through skipping C7 mutations in regions that are non-essential (57). Recent strategies include developing antisense ribonucleotides to target skipping of the mutated exon 73 or exon 80 of COL7A1 (58). This results in a portion of the mutated transcript being re-expressed and thus restoring partial C7 protein production. In vivo, these oligonucleotides can be injected into skin to restore anchoring fibril formation. Similarly, exon skipping was used to target exon 105, which also proved useful in an in vivo model (59). Another approach is trans-splicing, which involves combining the original mutated COL7A1 pre-mRNA and a synthetic 3′ trans-splicing molecule that results in a full-length, functional COL7A1 mRNA (60). Screening methods allow for assembly of trans-splicing constructs for particular types of RDEB-causing mutations (61).
Recently, there have been advances in gene-editing approaches that are capable of correcting endogenous mutations and restoring gene function for genetic disorders (62, 63, 64). For the treatment of epidermolysis bullosa, both TALEN and CRISPR/Cas approaches have been used to correct mutations in RDEB patient cells (37, 39). TALENs designed to correct a homozygous mutation in an RDEB patient fibroblast led to C7 expression with no off-target effects. Corrected fibroblasts were capable of expanding long term in culture, and being used to derive iPSC clones. A CRISPR/Cas gene-editing strategy was also used to correct a mutation in COL7A1 in cultured RDEB fibroblasts. These cells were then used to make iPSC lines capable of being differentiated into keratinocytes, mesenchymal lineages, and hematopoietic lineages. Off-target effects with this approach were limited. Similarly, an in vivo editing approach using CRISPR/Cas ribonucleoprotein was capable of correcting a COL7A1 mutation in a mouse model of RDEB by causing an in-frame deletion that restored function (65). Although there are limitations to these approaches, they appear to have less inherent risk than previous approaches for gene therapy. In addition, both the cost and time required to develop these tools are constantly shrinking. There are also a number of non-genetic approaches for treating RDEB, including aminoglycoside treatment, C7 protein therapy, and modulating RDEB disease using a pharmacological approach such as losartan treatment that may prove useful in complementing gene therapy and stem cell approaches (66, 67, 68, 69). In particular, C7 protein therapy has shown to be effective long-term via both injection and topical application, and may prove useful in a combinatorial approach for treating RDEB (67, 69).
Future strategies
Because the complex RDEB phenotype triggers a cascade of secondary pathological consequences, successful treatments will likely require combinatorial strategies (Figure 2). Although HCT for treating RDEB shows promise, HCT is a procedure with inherent risks including graft failure, graft-vs.-host disease, a transiently compromised immune system and side effects from the chemotherapy preparative regimen (16, 70). Although using HCT for treating RDEB has inherent risks, and not all patients treated have shown dramatic improvement, the potential for HCT or other stem cell therapies is promising and should be pursued and improved upon. Studying the biological mechanisms revealed by stem cell therapies like HCT and gene therapy will be valuable in guiding our future approaches. The subset or subsets of cells derived from HCT transplant that are effective in producing C7 and mediating wound healing have not been sufficiently characterized, although certain studies have given some insight into which cells may be responsible (21, 71). Identifying these subsets may help modify the transplant protocol or augment the therapy in ways that promote more C7 production in patients who are not good responders to HCT.

A vision of combinatorial therapy for treatment of RDEB. (80). Copyright © 2015 Massachusetts Medical Society. Reprinted with permission. RDEB, recessive dystrophic epidermolysis bullosa.
Furthermore, wound healing is a complex process, and it is unclear whether there are multiple cell types responsible for the important processes needed for sufficient, long-term improvement in RDEB skin, i.e., wound healing, C7 production, re-epithelialization, and long-term stability of the DEJ (7, 46, 72). There certainly may be immune cells that are important in the initial phase of wound healing and extracellular matrix production, but that do not contribute to long-term cellular populations in the skin (73, 74, 75). Conversely, there may be certain subsets of stem cells, such as MSCs or hematopoietic stem cells, that contribute to the cellular compartments of the wounded skin via differentiation or transdifferentiation, but that require certain conditions and time in order to provide meaningful therapeutic impact beyond the initial waves of differentiated immune cells (14, 18, 76). A thorough study of these aspects is needed to understand the complexity of using stem cell therapies to treat RDEB. Additional therapies, such as anti-fibrosis or anti-inflammatory medications, C7 protein therapy, and treatment with non-stem cell therapies such as genetically corrected keratinocyte sheets also have the potential to improve the overall well-being of RDEB patients (52, 68, 77).
There may be other, as-yet-unused cellular therapies that could be effective in treating certain conditions from which RDEB patients suffer, e.g., using limbal stem cells or corneal transplants to treat common and debilitating corneal abrasions (78, 79). Long-term studies of these therapies, including whether there is an alteration in the incidence of squamous cell carcinoma, also need to be conducted (11, 12).
Progress is being made, but there is much to be done to achieve a cure for RDEB (Figure 3). Future approaches should be forward thinking. For example, in regards to gene therapy, it may be safer and more beneficial in the long term to fix the gene on the inside than to provide an artificial, outside source of cells. From a stem cell aspect, giving stem cells that provide therapeutic benefit internally, such as hematopoietic stem cells, may provide a more systemic benefit than treatment with other cellular options. While difficult, fixing the inside—both the genetic component and the cellular component of RDEB—may be the best approach toward lasting benefits on the outside.

Advances in RDEB therapy over the last 15 years, at a cellular level (top) and genetic level (bottom). Cas, CRISPR-associated; CRISPR, clustered regularly interspaced short palindromic repeats; MSC, mesenchymal stem/stromal cell; RDEB, recessive dystrophic epidermolysis bullosa; SIN, self-inactivating; TALEN, transcription activator-like effector nuclease.
References
Chung HJ, Uitto J . Type VII collagen: the anchoring fibril protein at fault in dystrophic epidermolysis bullosa. Dermatol Clin 2010;28:93–105.
Kuttner V, Mack C, Rigbolt KT et al. Global remodelling of cellular microenvironment due to loss of collagen VII. Mol Syst Biol 2013;9:657.
Mack MR, Wendelschafer-Crabb G, McAdams BD, Hordinsky MK, Kennedy WR, Tolar J . Peripheral neuro-immune pathology in recessive dystrophic epidermolysis bullosa. J Invest Dermatol 2015;135:1193–1197.
Kirkorian AY, Weitz NA, Tlougan B, Morel KD . Evaluation of wound care options in patients with recessive dystrophic epidermolysis bullosa: a costly necessity. Pediatr Dermatol 2014;31:33–37.
Kon A, Takeda H, Ito N, Hanada K, Takagaki K . Tissue-specific downregulation of type VII collagen gene (COL7A1 transcription in cultured epidermal keratinocytes by ultraviolet A radiation (UVA) and UVA-inducible cytokines, with special reference to cutaneous photoaging. J Dermatol Sci 2005;1:S29–S35.
Nagy N, Almaani N, Tanaka A et al. HB-EGF induces COL7A1 expression in keratinocytes and fibroblasts: possible mechanism underlying allogeneic fibroblast therapy in recessive dystrophic epidermolysis bullosa. J Invest Dermatol 2011;131:1771–1774.
Nystrom A, Velati D, Mittapalli VR, Fritsch A, Kern JS, Bruckner-Tuderman L . Collagen VII plays a dual role in wound healing. J Clin Invest 2013;123:3498–3509.
Moon ES, Jung ST, Kim MS . Surgical treatment of pseudosyndactyly of children with epidermolysis bullosa: a case report. Hand Surg 2007;12:143–147.
Fine JD, Johnson LB, Weiner M et al. Eye involvement in inherited epidermolysis bullosa: experience of the National Epidermolysis Bullosa Registry. Am J Ophthalmol 2004;138:254–262.
Kirsner RS, Hu S . Recessive dystrophic epidermolysis bullosa and squamous-cell carcinoma: the role of type VII collagen. J Invest Dermatol 2007;127:2292.
Knaup J, Gruber C, Krammer B, Ziegler V, Bauer J, Verwanger T . TGFbeta-signaling in squamous cell carcinoma occurring in recessive dystrophic epidermolysis bullosa. Anal Cell Pathol 2011;34:339–353.
Mittapalli VR, Madl J, Loffek S et al. Injury-driven stiffening of the dermis expedites skin carcinoma progression. Cancer Res 2016;76:940–951.
Ng YZ, Pourreyron C, Salas-Alanis JC et al. Fibroblast-derived dermal matrix drives development of aggressive cutaneous squamous cell carcinoma in patients with recessive dystrophic epidermolysis bullosa. Cancer Res 2012;72:3522–3534.
Badiavas EV, Abedi M, Butmarc J, Falanga V, Quesenberry P . Participation of bone marrow derived cells in cutaneous wound healing. J Cell Physiol 2003;196:245–250.
Tolar J, Ishida-Yamamoto A, Riddle M et al. Amelioration of epidermolysis bullosa by transfer of wild-type bone marrow cells. Blood 2009;113:1167–1174.
Wagner JE, Ishida-Yamamoto A, McGrath JA et al. Bone marrow transplantation for recessive dystrophic epidermolysis bullosa. N Engl J Med 2010;363:629–639.
Tolar J, Wagner JE . Allogeneic blood and bone marrow cells for the treatment of severe epidermolysis bullosa: repair of the extracellular matrix. Lancet 2013;382:1214–1223.
Sasaki M, Abe R, Fujita Y, Ando S, Inokuma D, Shimizu H . Mesenchymal stem cells are recruited into wounded skin and contribute to wound repair by transdifferentiation into multiple skin cell type. J Immunol 2008;180:2581–2587.
Perdoni C, McGrath JA, Tolar J . Preconditioning of mesenchymal stem cells for improved transplantation efficacy in recessive dystrophic epidermolysis bullosa. Stem Cell Res Ther 2014;5:121.
Petrof G, Lwin SM, Martinez-Queipo M et al. Potential of systemic allogeneic mesenchymal stromal cell therapy for children with recessive dystrophic epidermolysis bullosa. J Invest Dermatol 2015;135:2319–2321.
Tamai K, Yamazaki T, Chino T et al. PDGFRalpha-positive cells in bone marrow are mobilized by high mobility group box 1 (HMGB1) to regenerate injured epithelia. Proc Natl Acad Sci USA 2011;108:6609–6614.
Iinuma S, Aikawa E, Tamai K et al. Transplanted bone marrow-derived circulating PDGFRalpha+ cells restore type VII collagen in recessive dystrophic epidermolysis bullosa mouse skin graft. J Immunol 2015;194:1996–2003.
Green H, Kehinde O, Thomas J . Growth of cultured human epidermal cells into multiple epithelia suitable for grafting. Proc Natl Acad Sci USA 1979;76:5665–5668.
Barrandon Y, Green H . Three clonal types of keratinocyte with different capacities for multiplication. Proc Natl Acad Sci USA 1987;84:2302–2306.
Takahashi K, Yamanaka S . Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006;126:663–676.
Yu J, Vodyanik MA, Smuga-Otto K et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007;318:1917–1920.
Hanna J, Wernig M, Markoulaki S et al. Treatment of sickle cell anemia mouse model with iPS cells generated from autologous skin. Science 2007;318:1920–1923.
Tedesco FS, Gerli MF, Perani L et al. Transplantation of genetically corrected human iPSC-derived progenitors in mice with limb-girdle muscular dystrophy. Sci Transl Med 2012;4:140ra89.
Mandai M, Watanabe A, Kurimoto Y et al. Autologous induced stem-cell-derived retinal cells for macular degeneration. N Engl J Med 2017;376:1038–1046.
Araki R, Uda M, Hoki Y et al. Negligible immunogenicity of terminally differentiated cells derived from induced pluripotent or embryonic stem cells. Nature 2013;494:100–104.
Tolar J, Xia L, Riddle MJ et al. Induced pluripotent stem cells from individuals with recessive dystrophic epidermolysis bullosa. J Invest Dermatol 2011;131:848–856.
Tolar J, Xia L, Lees CJ et al. Keratinocytes from induced pluripotent stem cells in junctional epidermolysis bullosa. J Invest Dermatol 2013;133:562–565.
Itoh M, Kawagoe S, Tamai K, Okano HJ, Nakagawa H . Integration-free T cell-derived human induced pluripotent stem cells (iPSCs) from a patient with recessive dystrophic epidermolysis bullosa (RDEB) carrying two compound heterozygous mutations in the COL7A1 gene. Stem Cell Res 2016;17:32–35.
Tolar J, McGrath JA, Xia L et al. Patient-specific naturally gene-reverted induced pluripotent stem cells in recessive dystrophic epidermolysis bullosa. J Invest Dermatol 2014;134:1246–1254.
Umegaki-Arao N, Pasmooij AM, Itoh M et al. Induced pluripotent stem cells from human revertant keratinocytes for the treatment of epidermolysis bullosa. Sci Transl Med 2014;6:264ra164.
Chang CJ, Bouhassira EE . Zinc-finger nuclease-mediated correction of alpha-thalassemia in iPS cells. Blood 2012;120:3906–3914.
Osborn MJ, Starker CG, McElroy AN et al. TALEN-based gene correction for epidermolysis bullosa. Mol Ther 2013;21:1151–1159.
Sebastiano V, Zhen HH, Haddad B et al. Human COL7A1-corrected induced pluripotent stem cells for the treatment of recessive dystrophic epidermolysis bullosa. Sci Transl Med 2014;6:264ra163.
Webber BR, Osborn MJ, McElroy AN et al. CRISPR/Cas9-based genetic correction for recessive dystrophic epidermolysis bullosa. NPJ Regen Med 2016 (e-pub ahead of print 8 December 2016; doi:10.1038/npjregenmed.2016.14).
Tolar J, McGrath JA . The three-body problem of therapy with induced pluripotent stem cells. Genome Med 2015;7:15.
Webber BR, Tolar J . From marrow to matrix: novel gene and cell therapies for epidermolysis bullosa. Mol Ther 2015;23:987–992.
Inoue T, Kulkeaw K, Okayama S, Tani K, Sugiyama D . Variation in mesodermal and hematopoietic potential of adult skin-derived induced pluripotent stem cell lines in mice. Stem Cell Rev 2011;7:958–968.
Suzuki N, Yamazaki S, Yamaguchi T et al. Generation of engraftable hematopoietic stem cells from induced pluripotent stem cells by way of teratoma formation. Mol Ther 2013;21:1424–1431.
Tolar J, Park IH, Xia L et al. Hematopoietic differentiation of induced pluripotent stem cells from patients with mucopolysaccharidosis type I (Hurler syndrome). Blood 2011;117:839–847.
Sugimura R, Jha DK, Han A et al. Haematopoietic stem and progenitor cells from human pluripotent stem cells. Nature 2017;545:432–438.
Petrof G, Martinez-Queipo M, Mellerio JE, Kemp P, McGrath JA . Fibroblast cell therapy enhances initial healing in recessive dystrophic epidermolysis bullosa wounds: results of a randomized, vehicle-controlled trial. Br J Dermatol 2013;169:1025–1033.
Itoh M, Umegaki-Arao N, Guo Z, Liu L, Higgins CA, Christiano AM . Generation of 3D skin equivalents fully reconstituted from human induced pluripotent stem cells (iPSCs). PLoS ONE 2013;8:e77673.
Awe JP, Lee PC, Ramathal C et al. Generation and characterization of transgene-free human induced pluripotent stem cells and conversion to putative clinical-grade status. Stem Cell Res Ther 2013;4:87.
Jackow J, Titeux M, Portier S et al. Gene-corrected fibroblast therapy for recessive dystrophic epidermolysis bullosa using a self-inactivating COL7A1 retroviral vector. J Invest Dermatol 2016;136:1346–1354.
Gache Y, Baldeschi C, Del Rio M et al. Construction of skin equivalents for gene therapy of recessive dystrophic epidermolysis bullosa. Hum Gene Ther 2004;15:921–933.
Titeux M, Pendaries V, Zanta-Boussif MA et al. SIN retroviral vectors expressing COL7A1 under human promoters for ex vivo gene therapy of recessive dystrophic epidermolysis bullosa. Mol Ther 2010;18:1509–1518.
Siprashvili Z, Nguyen NT, Gorell ES et al. Safety and wound outcomes following genetically corrected autologous epidermal grafts in patients with recessive dystrophic epidermolysis bullosa. JAMA 2016;316:1808–1817.
Chen M, Kasahara N, Keene DR et al. Restoration of type VII collagen expression and function in dystrophic epidermolysis bullosa. Nat Genet 2002;32:670–675.
Georgiadis C, Syed F, Petrova A et al. Lentiviral engineered fibroblasts expressing codon-optimized COL7A1 restore anchoring fibrils in RDEB. J Invest Dermatol 2016;136:284–292.
Cutlar L, Gao Y, Aied A et al. A knot polymer mediated non-viral gene transfection for skin cells. Biomater Sci 2016;4:92–95.
Latella MC, Cocchiarella F, De Rosa L et al. Correction of recessive dystrophic epidermolysis bullosa by transposon-mediated integration of COL7A1 in transplantable patient-derived primary keratinocytes. J Invest Dermatol 2017;137:836–844.
Goto M, Sawamura D, Nishie W et al. Targeted skipping of a single exon harboring a premature termination codon mutation: implications and potential for gene correction therapy for selective dystrophic epidermolysis bullosa patients. J Invest Dermatol 2006;126:2614–2620.
Turczynski S, Titeux M, Tonasso L, Decha A, Ishida-Yamamoto A, Hovnanian A . Targeted exon skipping restores type VII collagen expression and anchoring fibril formation in an in vivo RDEB model. J Invest Dermatol 2016;136:2387–2395.
Bremer J, Bornert O, Nystrom A et al. Antisense oligonucleotide-mediated exon skipping as a systemic therapeutic approach for recessive dystrophic epidermolysis bullosa. Mol Ther Nucleic Acids 2016;5:e379.
Murauer EM, Gache Y, Gratz IK et al. Functional correction of type VII collagen expression in dystrophic epidermolysis bullosa. J Invest Dermatol 2011;131:74–83.
Tockner B, Kocher T, Hainzl S et al. Construction and validation of an RNA trans-splicing molecule suitable to repair a large number of COL7A1 mutations. Gene Ther 2016;23:775–784.
Carroll D . Genome engineering with zinc-finger nucleases. Genetics 2011;188:773–782.
Gaj T, Gersbach CA, Barbas CF 3rd . ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol 2013;31:397–405.
Reyon D, Tsai SQ, Khayter C, Foden JA, Sander JD, Joung JK . FLASH assembly of TALENs for high-throughput genome editing. Nat Biotechnol 2012;30:460–465.
Wu W, Lu Z, Li F et al. Efficient in vivo gene editing using ribonucleoproteins in skin stem cells of recessive dystrophic epidermolysis bullosa mouse model. Proc Natl Acad Sci USA 2017;114:1660–1665.
Cogan J, Weinstein J, Wang X et al. Aminoglycosides restore full-length type VII collagen by overcoming premature termination codons: therapeutic implications for dystrophic epidermolysis bullosa. Mol Ther 2014;22:1741–1752.
Hou Y, Guey LT, Wu T et al. Intravenously administered recombinant human type VII collagen derived from Chinese hamster ovary cells reverses the disease phenotype in recessive dystrophic epidermolysis bullosa mice. J Invest Dermatol 2015;135:3060–3067.
Nystrom A, Thriene K, Mittapalli V et al. Losartan ameliorates dystrophic epidermolysis bullosa and uncovers new disease mechanisms. EMBO Mol Med 2015;7:1211–1228.
Wang X, Ghasri P, Amir M et al. Topical application of recombinant type VII collagen incorporates into the dermal-epidermal junction and promotes wound closure. Mol Ther 2013;21:1335–1344.
Geyer MB, Radhakrishnan K, Giller R et al. Reduced toxicity conditioning and allogeneic hematopoietic progenitor cell transplantation for recessive dystrophic epidermolysis bullosa. J Pediatr 2015;167:e1.
Liao Y, Itoh M, Yang A et al. Human cord blood-derived unrestricted somatic stem cells promote wound healing and have therapeutic potential for patients with recessive dystrophic epidermolysis bullosa. Cell Transplant 2014;23:303–317.
Conget P, Rodriguez F, Kramer S et al. Replenishment of type VII collagen and re-epithelialization of chronically ulcerated skin after intradermal administration of allogeneic mesenchymal stromal cells in two patients with recessive dystrophic epidermolysis bullosa. Cytotherapy 2010;12:429–431.
Abe R, Donnelly SC, Peng T, Bucala R, Metz CN . Peripheral blood fibrocytes: differentiation pathway and migration to wound sites. J Immunol 2001;166:7556–7562.
Ito M, Liu Y, Yang Z et al. Stem cells in the hair follicle bulge contribute to wound repair but not to homeostasis of the epidermis. Nat Med 2005;11:1351–1354.
Leoni G, Neumann PA, Sumagin R, Denning TL, Nusrat A . Wound repair: role of immune-epithelial interactions. Mucosal Immunol 2015;8:959–968.
Inokuma D, Abe R, Fujita Y et al. CTACK/CCL27 accelerates skin regeneration via accumulation of bone marrow-derived keratinocytes. Stem Cells 2006;24:2810–2816.
Woodley DT, Wang X, Amir M et al. Intravenously injected recombinant human type VII collagen homes to skin wounds and restores skin integrity of dystrophic epidermolysis bullosa. J Invest Dermatol 2013;133:1910–1913.
Hayashi R, Ishikawa Y, Ito M et al. Generation of corneal epithelial cells from induced pluripotent stem cells derived from human dermal fibroblast and corneal limbal epithelium. PLoS ONE 2012;7:e45435.
Rama P, Matuska S, Paganoni G, Spinelli A, De Luca M, Pellegrini G . Limbal stem-cell therapy and long-term corneal regeneration. N Engl J Med 2010;363:147–155.
Tolar J, Wagner JE . A biologic Velcro patch. N Engl J Med 2015;372:382–384.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Statement of financial support
We gratefully acknowledge the support from NIH grant NHLBI R01 AR063070 (J.T.) and NIH Training Grant T32 GM113846 (M.V.O.).
Rights and permissions
About this article

Cite this article
Vanden Oever, M., Twaroski, K., Osborn, M. et al. Inside out: regenerative medicine for recessive dystrophic epidermolysis bullosa. Pediatr Res 83, 318–324 (2018). https://doi.org/10.1038/pr.2017.244
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/pr.2017.244
This article is cited by
-
Gene-edited cells: novel allogeneic gene/cell therapy for epidermolysis bullosa
Journal of Applied Genetics (2024)
-
Investigational Treatments for Epidermolysis Bullosa
American Journal of Clinical Dermatology (2021)
-
Pathogenetic Therapy of Epidermolysis Bullosa: Current State and Prospects
Bulletin of Experimental Biology and Medicine (2021)
