
Abstract
Background
Necrotizing enterocolitis (NEC) is a devastating neonatal disease characterized by intestinal necrosis. Hypoxia-inducible factor-1α (HIF-1α) has a critical role in cellular oxygen homeostasis. Here, we hypothesized that prolyl hydroxylase (PHD) inhibition, which stabilizes HIF-1α, protects against NEC by promoting intestinal endothelial cell proliferation and improving intestinal microvascular integrity via vascular endothelial growth factor (VEGF) signaling.
Methods
To assess the role of PHD inhibition in a neonatal mouse NEC model, we administered dimethyloxalylglycine (DMOG) or vehicle to pups before or during the NEC protocol, and determined mortality and incidence of severe intestinal injury. We assessed intestinal VEGF by western blot analysis and quantified endothelial cell and epithelial cell proliferation following immunofluorescence.
Results
DMOG decreased mortality and incidence of severe NEC, increased intestinal VEGF expression, and increased intestinal villus endothelial and epithelial cell proliferation in experimental NEC. Inhibiting VEGFR2 signaling eliminated DMOG’s protective effect on intestinal injury severity, survival, and endothelial cell proliferation while sparing DMOG’s protective effect on intestinal epithelial cell proliferation.
Conclusion
DMOG upregulates intestinal VEGF, promotes endothelial cell proliferation, and protects against intestinal injury and mortality in experimental NEC in a VEGFR2 dependent manner. DMOG’s protective effect on the neonatal intestinal mucosa may be mediated via VEGFR2 dependent improvement of the intestinal microvasculature.
Similar content being viewed by others

Main
Necrotizing enterocolitis (NEC), a devastating disease of the neonatal intestine, affects 7–10% of very low birth weight infants. It is characterized by variable degrees of intestinal tissue necrosis (1). NEC pathogenesis is multifactorial (2, 3). Translocation of bacteria across a weakened intestinal barrier in association with impaired intestinal mucosal immunity leads to an exaggerated inflammatory response, which contributes to a vicious cycle of inflammation and secondary mucosal epithelial injury (2, 3). Mortality is reported as high as 30% (ref. 4), and survivors suffer from significant long-term morbidities including short bowel syndrome, and significant neurodevelopmental delays (5). No specific therapies are currently available for NEC, with treatment limited to supportive care and surgery in severely affected infants.
The intestinal microvasculature may have an important role in NEC (6, 7). We recently found that vascular endothelial growth factor (VEGF), a major regulator of angiogenesis, and its main receptor VEGF receptor 2 (VEGFR2) are strongly expressed during late fetal life (7) and decreased in experimental NEC (6). Furthermore, VEGF is decreased in human NEC (6). Inhibition of VEGFR2 kinase activity decreased villous endothelial cell proliferation and increased mortality and severe NEC incidence in neonatal mice (7). VEGF is regulated by the transcription factor hypoxia-inducible factor-1 (HIF-1), but the role of HIF-1 in NEC remains unknown.
HIF-1, the master regulator of the cellular response to hypoxia and ischemia, is a highly conserved transcription factor present in all mammals (8, 9). HIF-1α mediates angiogenesis in response to hypoxic injury by regulating expression of growth factors including VEGF-A, which stimulates endothelial cell proliferation, promotes migration of endothelial cells and vascular smooth muscle cells, and inhibits apoptosis (10, 11). The intestinal mucosa experiences profound fluctuations in the blood flow and oxygenation during normal physiological conditions, and, accordingly, intestinal epithelial cells (IECs) are uniquely resistant to hypoxia (12). IECs cope with hypoxia through basal regulation of HIF (12). Oxygen-dependent hydroxylation of HIF-1α by prolyl hydroxylase domain enzymes (PHDs) signals the Von Hippel Lindau protein to bind and target HIF-1α subunits for proteasomal degradation, thereby regulating HIF-1α abundance (13). In hypoxia, HIF-1α is stabilized, permitting heterodimerization with HIF-1β. The active HIF-1 heterodimer translocates into the nucleus and binds to HIF response elements present in the promoter of HIF target genes, thereby activating transcription of genes that regulate oxygen homeostasis, including VEGF-A (11). Although studies have shown increased HIF-1α expression in the intestine during experimental NEC (14), whether this represents a protective response to intestinal injury or contributes to injury development remains unknown.
Here, we hypothesized that HIF-1α stabilization protects against tissue injury in experimental NEC via upregulation of VEGFR2 signaling, thus improving intestinal microvascular integrity. In this study, using dimethyloxalylglycine (DMOG), a PHD inhibitor that prevents HIF-1α degradation in normoxia (15), we investigated whether PHD inhibition ameliorates intestinal tissue injury in our well-characterized neonatal mouse NEC model (16). We further evaluated whether DMOG’s protective effects on mortality and severity of intestinal injury in experimental NEC are dependent on VEGFR2 signaling. To elucidate the mechanism by which PHD inhibition protects against NEC, we examined DMOG’s effects on intestinal VEGF expression and intestinal villus endothelial cell and IEC proliferation, and evaluated whether DMOG-mediated preservation of IEC and endothelial cell proliferation in experimental NEC is dependent on VEGFR2 signaling in the neonatal mouse intestine.
Methods
Materials
C57BL/6 mice were purchased from Jackson Laboratory (Bar Harbor, ME). All animal breeding and procedures were approved by the Institutional Animal Care and Use Committee of Stanley Manne Children’s Research Institute/Northwestern University. Animals received humane care in compliance with the Guide for the Care and Use of Laboratory Animals (NIH Publication No. 85Y23, revised 1996). Anti-CD31 (#ab28364) and anti-5-bromo-2'-deoxyuridine (BrdU; #ab6326) antibodies were purchased from Abcam (Cambridge, MA), anti-VEGF-A (#sc-7269) antibodies from Santa Cruz Biotechnology (Santa Cruz, CA), and horseradish peroxidase-conjugated antibodies (#A11037 and #A11007) from Life Technologies (Grand Island, NY). DMOG was purchased from Sigma-Aldrich (St Louis, MO) and Ki8751 from R&D Systems (Minneapolis, MN).
Animal Experiments
To study the effect of PHD inhibition on NEC incidence, litters of newborn (P0) pups were injected with PHD inhibitor DMOG 40 mg/kg intraperitoneally (IP; in 30 μl phosphate-buffered saline (PBS)/1% carboxymethyl cellulose (CMC)) or vehicle (PBS/1% CMC) and 18 h later subjected to our NEC protocol (16). We induced NEC via a 72-h protocol consisting of inoculation with standardized murine adult commensal bacteria (108 colony-forming unit) and 5 mg/kg lipopolysaccharide to perturb normal intestinal flora colonization, Esbilac formula-feeding every 3 h (200 ml/kg/day), and exposure to asphyxia (60 s in 100% N2) followed by cold stress (10 min in 4 °C) twice daily. This protocol induces within 72 h a range of intestinal injury from mild (epithelial) to severe (transmural) resembling human NEC. During this 72-h experimental period, animals were continuously monitored for clinical signs of NEC (severe abdominal distension, lethargy, and apnea). Pups surviving for 72 h without signs of distress and pups who developed signs of clinical distress or imminent death were killed by decapitation and time of death was recorded. Whole intestinal tissues were collected and hematoxylin and eosin-stained slides evaluated. The most severely affected area was scored by an investigator unaware of the group assignment, using a pre-established scoring system (16). Severe NEC was defined as histological score ⩾2, encompassing mid-villous necrosis (score −2), complete villous necrosis (score −3), or transmural necrosis (score −4), corresponding to clinically significant NEC. Mild epithelial sloughing (score −1) is nonspecific and may not represent illness. In all, 6/50 vehicle-treated pups and 3/54 DMOG-treated pups were found dead while not under direct observation and were excluded from histological analysis because of concern that autolysis following a brief delay between death and tissue processing would compromise intestinal injury assessment validity.
To study DMOG’s route of delivery and timing of administration, the following three groups were used: Group 1 (DMOG-gavage) received prophylactic DMOG 40 mg/kg by orogastric gavage and 18 h later was subjected to the NEC protocol; Group 2 (IP-DMOG-therapeutic) received 40 mg/kg DMOG IP at 18 h of NEC induction; Group 3 received IP vehicle only at 18 h of NEC induction.
To study whether DMOG’s protective effect persists when VEGFR2 signaling is inhibited, pups received either IP DMOG (40 mg/kg) or vehicle. A subset of pups also received the selective VEGFR2 inhibitor Ki8751 (0.75 mg/kg, IP), 1 h after DMOG injection. Ki8751 is over 40-fold more selective for VEGFR2 than c-Kit, PDGFRα, and FGFR-2, and has little activity on EGFR, HGFR, and insulin receptor (17). Eighteen hours after DMOG or vehicle, pups were exposed to a NEC protocol or were left with dams for 72 h.
To study the effect of DMOG on VEGF in the mouse intestine, P8 littermates were randomized to receive either DMOG (40 mg/kg, IP) or vehicle. Forty-eight hours later, collected small intestines were analyzed for VEGF protein expression using western blot analysis.
For endothelial cell proliferation assessment, P0 pups were divided into four groups: DF pups injected with DMOG (40 mg/kg, IP); DF pups injected with vehicle only; pups injected with DMOG and subjected 18 h later to our NEC protocol; pups injected with vehicle and subjected 18 h later to our NEC protocol. After 24 h, the pups were killed and intestinal tissues were collected for immunofluorescence staining. DF pups were killed ~42 h after injection with DMOG or vehicle (to correspond to 18 h pretreatment with DMOG and 24 h NEC induction in littermates exposed to the NEC protocol). To label proliferating cells, BrdU (0.3 mg IP) was injected 4 h before being killed. To study whether DMOG promotes endothelial cell proliferation via VEGFR2, littermates were injected with DMOG or vehicle only. A subset of pups received Ki8751 (IP 0.75 mg/kg). BrdU was administered and intestinal tissues collected. All time points described refer to hours after NEC induction in experimental pups and their littermate DF controls unless otherwise specified.
Immunofluorescence Staining
Five-μm-thick formalin-fixed paraffin-embedded intestinal tissue sections were stained using standard immunofluorescent methods for CD31 and BrdU. Slides were incubated with primary antibodies (anti-CD31 (1:50), anti-BrdU (1:50)) overnight at 4 °C. Washed slides were incubated for 1 h with fluorescent dye-conjugated secondary antibody (goat anti-rabbit secondary antibody (1:500), goat anti-rat secondary antibody (1:1,000)) for 1 h at room temperature. Slides were mounted in Prolong Gold antifade reagent with 4', 6-diamidino-2-phenylindole (Life Technologies) and images were captured using a mac_aq id="Q5"> (Leica, Buffalo Grove, IL, USA) DMR-HC upright microscope. Proliferating endothelial cells (co-staining for CD31 and BrdU) were assessed in 5–10 × 20 power fields with five mice per group, by a blinded investigator. Negative controls in which the primary antibody had been omitted were run concomitantly and did not show staining. The number of proliferating BrdU+ IECs per crypt was also assessed in five to seven × 20 power fields with five to seven mice per group.
Western Blotting
Snap-frozen small intestines were homogenized in lysis buffer (10 mM Tris-Cl pH 7.6, 150 mM NaCl, 5 mM EDTA, 1 mM phenylmethanesulfonyl fluoride, 1 mM dithiothreitol, 0.25% Nonidet P-40, and Complete Mini Tablet Protease Inhibitor Cocktail from Roche Biochemical Reagents (Sigma-Aldrich)) and lysate protein concentration determined (Bradford method). Fifty micrograms of protein were run on 10 or 15% sodium dodecyl sulfate-polyacrylamide gel electrophoresis gels and transferred onto polyvinylidene fluoride membranes. Membranes were blocked (milk in Tris-buffered saline-Tween 20, 1 h), incubated with primary antibodies overnight (4 °C), and then with horseradish peroxidase-conjugated secondary antibodies (1 h, room temperature). Membranes were developed using the Pierce ECL method. Relative protein expression was determined using densitometry and normalized to β-actin.
Statistical Analysis
ANOVA was performed when more than two groups were compared. Two-tailed unpaired Student’s t-test was used for comparison between the two groups. Results were expressed as mean+SEM unless otherwise specified. Differences in severe NEC incidence (score ⩾2) were evaluated using χ2-analysis. Animal survival data were analyzed by log-rank test. Differences were considered statistically significant when P values were <0.05.
Results
DMOG Decreases Mortality and the Incidence of Severe Intestinal Injury in a Murine Neonatal NEC Model
To determine whether HIF-1α stabilization has a role in intestinal injury in NEC, we injected pups IP with the PHD inhibitor DMOG (40 mg/kg) or vehicle alone 18 h before NEC induction (Figure 1a). The treatment dose of 40 mg/kg was chosen based on several in vivo studies showing that DMOG at this dose increases HIF-1α gene expression and protects against cerebral and myocardial ischemic injury in several rodent models (18, 19). We found that DMOG significantly decreased the mortality of neonatal mice exposed to the NEC protocol by 2.16-fold (DMOG: 12/54 vs. 24/50, P<0.005, Figure 1b). Furthermore, when pups were injected with DMOG before NEC induction, the incidence of severe intestinal injury (histological injury score ⩾2) was decreased by 1.83-fold compared with those pups injected with vehicle only (DMOG: 18/51 vs. 28/44, χ2=7.6, P<0.01, Figure 1c).

DMOG decreases 72 h mortality and the incidence of severe intestinal injury in experimental NEC. (a–c) Newborn mice were injected with DMOG (40 mg/kg, IP) or vehicle 18 h before a NEC protocol. Mice were killed when presenting signs of distress or at 72 h of the NEC protocol and whole intestines processed for histology. A timeline of the experiment (a), survival curves (b), and histology scores (c) are presented. Survival was correlated with severity of intestinal injury in both groups studied (d). Data represent three independent experiments combined. (e,f) To study the impact of DMOG’s delivery route and timing of administration, littermates were divided into three groups: Group 1 received prophylactic DMOG (40 mg/kg) by orogastric gavage followed 18 h later by NEC induction; Group 2 (DMOG-IP therapeutic) received 40 mg/kg DMOG or vehicle via IP injection at 18 h of NEC induction; Group 3 received vehicle (IP) at 18 h of NEC induction. All three groups were exposed to a 72 h NEC protocol. Survival curves (e) and histology scores (f) are presented. *P<0.05, **P<0.005, ***P<0.001, ****P<0.0001. Data represent two independent experiments combined. DMOG, dimethyloxalylglycine; IP, intraperitoneally; NEC, Necrotizing enterocolitis.
As expected, mortality was correlated to intestinal injury severity. We found that 83% of pups with histology scores of 0–1 survived the 72-h experimental period vs. 68% of pups with scores of 2 vs. 42% of pups with scores of 3 vs. 12.5% of pups with scores of 4 (P<0.0001; Figure 1d). There was a 6.6-fold higher mortality in pups with the highest intestinal injury scores (scores of 4) when compared with pups with mild or no intestinal injury (scores of 0–1; χ2=25.3, P<0.0001). However, there was no statistically significant difference in mortality between DMOG-treated pups and vehicle-treated pups at similar histology scores.
To determine whether timing or route of administration affected DMOG’s protective effect, littermates were randomly assigned to the following three groups: (i) 40 mg/kg DMOG prophylaxis by orogastric gavage 18 h before NEC induction; (ii) DMOG (40 mg/kg, IP) at 18 h of NEC induction; or (iii) vehicle only by IP injection (Figure 1a). We found that DMOG’s protective effect on survival and intestinal injury in experimental NEC was also present when administered enterally before NEC induction. When compared with controls, enterally administered DMOG significantly decreased NEC-associated mortality by 1.87-fold (11/27 vs. 19/25, P<0.05; Figure 1e) and decreased the incidence of severe NEC (histological injury score ⩾2) by 2.89-fold (7/27 vs. 18/24, χ2=3.5, P<0.001; Figure 1f). Administration of DMOG at 18 h of NEC induction was still associated with a decreased incidence of severe NEC 3.5-fold compared with vehicle-treated littermates (6/28 vs. 18/24, χ2=14.92, P<0.0001; Figure 1f), although no significant improvement in mortality was noted (18/29 (62%) vs. 19/25 (76%), not significant; Figure 1e).
DMOG Increases Intestinal VEGF Expression in Pups
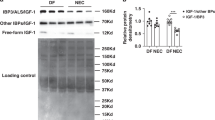
To determine whether DMOG affects intestinal VEGF expression, dam-fed (DF) pups were injected with 40 mg/kg IP DMOG or vehicle only and intestinal tissues were collected 48 h later. VEGF expression was assessed using western blot analysis of whole intestinal tissue lysates. We found that injection with DMOG significantly increased VEGF protein expression in the intestine of DF pups (Figure 2a).

VEGF-A expression is increased in the mouse intestine following injection with DMOG. Intestinal tissues from 8-day-old pups were obtained 48 h after DMOG (40 mg/kg, IP) or vehicle only (n=3/group). VEGF-A protein was analyzed by western blot analysis of small intestinal tissue lysates (a). Band densitometry values normalized to β-actin are presented in b. **P<0.005. DMOG, dimethyloxalylglycine; IP, intraperitoneally; VEGF, vascular endothelial growth factor.
Inhibition of VEGFR2 Signaling Abrogates DMOG’s Protective Effect on Mortality and Intestinal Injury
To determine whether DMOG’s protective effect on mortality and intestinal injury in NEC was dependent on VEGFR2 signaling, newborn pups were injected with DMOG (IP 40 mg/kg) or vehicle only (PBS-CMC; littermate controls). A subset of pups in each group was also injected with Ki8751 (0.75 mg/kg IP), a selective VEGFR2 tyrosine kinase inhibitor. Pups in each group were subjected to the NEC model, with a small subset of pups in each group remaining with their dams. Survival over the 72 h NEC model and histological injury analyses were performed. As expected, DMOG prophylaxis significantly decreased the mortality of neonatal mice exposed to the NEC protocol compared with vehicle-treated littermates (11/16 vs. 15/17, P<0.05, Figure 3a). We previously showed that inhibition of VEGFR2 tyrosine kinase activity by Ki8751 increased the incidence of severe NEC and mortality (7). Ki8751 completely abrogated DMOG’s protective effect on 72-h mortality (17/17 vs. 11/16, P<0.05), to levels similar to Ki8751 treatment alone (17/17 vs. 17/17, NS; Figure 3a). Similarly, DMOG’s protective effect on severe intestinal injury was eliminated by VEGFR2 inhibition (14/16 vs. 3/16, χ2=15.2, P<0.0001; Figure 3b) to a level similar to Ki8751 alone (13/15; Figure 3b). However, Ki8751 did not affect the 72-h survival in DF pups as 90% (9/10) of DF Ki8751-treated pups survived the 72-h observation (vs. 9/10 of DF DMOG/Ki8751-treated pups and 8/8 of DMOG-treated pups, NS; Figure 3a). As expected, none of the DMOG-treated DF pups (0/8) had severe intestinal injury on histological examination. Interestingly, 3/9 Ki8751-treated DF pups and 4/8 DMOG-Ki8751-treated DF pups had severe intestinal injury following Ki8751 treatment alone (Figure 3c), consistent with our previous findings that VEGFR2 inhibition predisposes to intestinal injury in neonatal mice (7).

Inhibition of VEGFR2 signaling abrogates DMOG’s protective effect on mortality and intestinal injury. Newborn pups received either DMOG (40 mg/kg, IP) or vehicle. A subset of pups in each group also received Ki8751 (0.75 mg/kg, IP) or vehicle. Eighteen hours later, pups were exposed to the NEC protocol or were left with their dams. Mice were killed when presenting signs of distress or at 72 h (Figure 1a) and whole intestines processed for histology. Survival curves (a) and histology scores are presented representing NEC (b) and DF (c). ***P<0.001, ****P<0.0001 DF, dam-fed; DMOG, dimethyloxalylglycine; IP, intraperitoneally; NEC, necrotizing enterocolitis; VEGFR2, vascular endothelial growth factor receptor 2.
Endothelial Cell and Epithelial Cell Proliferation are Decreased in the Intestinal Villi of Neonatal Pups During NEC Induction, and Both Effects are Mitigated by DMOG Administration
To determine whether DMOG preserves intestinal endothelial cell and epithelial cell proliferation during NEC, P0 mouse pups were injected with DMOG (IP 40 mg/kg) or vehicle only (littermate controls). Eighteen hours later, half the pups in each group underwent NEC induction and the other half were left with the dams and DF. Twenty-four hours later, small intestinal tissues were collected (4h after BrdU injection) and tissue sections immunostained for BrdU and CD31. Intestinal endothelial cell proliferation was decreased in vehicle-treated NEC pups compared with that in vehicle-treated DF littermate controls (7.8+0.9 vs. 19.0+1.2 cells/field, P<0.0001), an effect that was mitigated by DMOG administration (DMOG 11.5 vs. VT-NEC 7.8+0.9 cells/field, P<0.005; Figure 4b). In addition, in DF pups, DMOG increased villous endothelial cell proliferation compared with vehicle-treated littermate controls (26.8+1.8 vs. 19.7+2.0 cells/high power fields, P<0.05; Figure 4a–c).

Inhibiting VEGFR2 signaling abrogates DMOG’s protective effect on villous endothelial cell proliferation in DF and NEC-exposed newborn mice. P0–1 littermates received either DMOG (40 mg/kg, IP) or vehicle. A subset in each group also received Ki8751 (0.75 mg/kg, IP). Eighteen hours later, pups were either exposed to the NEC protocol for 24 h or were left with their dams and DF. Pups were injected with BrdU (0.3 mg IP) 4 h before being killed. Immunofluorescence staining for BrdU (red) and CD31 (green) was performed on small intestinal tissues (a). Proliferating endothelial cells (CD31+BrdU+ cells) in the intestinal villi were counted by an investigator unaware of group assignment. Five to seven fields were examined per section with three to five mice per group; bar=50 μm. The mean numbers of proliferating endothelial cells per field (±SEM) are presented (b,c). NEC (b) and DF (c). *P<0.05, **P<0.005, ***P<0.0001. BrdU, 5-bromo-2'-deoxyuridine; DF, dam-fed; DMOG, dimethyloxalylglycine; IP, intraperitoneally; NEC, necrotizing enterocolitis; VEGFR2, vascular endothelial growth factor receptor 2.
We also found that the number of proliferating epithelial cells per intestinal crypt was decreased in vehicle-treated NEC pups compared with that in vehicle-treated DF littermate controls (3.1+0.24 vs. 6.17+0.35, P<0.0001; Figure 5). Treatment with DMOG partially rescued IEC proliferation compared with vehicle-treated littermates (4.9+0.50 vs. 3.1+0.24, P=0.001; Figure 5).

DMOG partially rescues intestinal epithelial cell proliferation in experimental NEC in a VEGFR2-independent manner. Littermates received either DMOG (40 mg/kg, IP) or vehicle. A subset in each group also received Ki8751 (0.75 mg/kg, IP). Eighteen hours after DMOG or vehicle injection, the pups were submitted to a NEC protocol for 24 h. DF vehicle-treated pups were studied as controls. Pups were injected with BrdU (0.3 mg IP) 4 h before being killed. Small intestinal tissues were processed for BrdU immunofluorescence staining. Proliferating BrdU+ intestinal epithelial cells per crypt were quantified. Five to seven fields were examined per section with five to seven mice per group. The mean numbers of proliferating epithelial cells per crypt (±SEM) are presented. ***P<0.0001. BrdU, 5-bromo-2'-deoxyuridine; DF, dam-fed; DMOG, dimethyloxalylglycine; IP, intraperitoneally; NEC, necrotizing enterocolitis VEGFR2, vascular endothelial growth factor.
Inhibition of VEGFR2 Signaling Abrogates DMOG’s Protective Effect on Villous Endothelial Cell Proliferation but does not Affect DMOG’s Protective Effect on IEC Proliferation in a Neonatal Mouse Model of NEC
To determine whether increased intestinal endothelial cell proliferation in DMOG-treated pups is mediated via VEGFR2 signaling, DF mouse pups were injected with DMOG (IP 40 mg/kg) or vehicle only (PBS-CMC; littermate controls). Half the pups in each group were also injected with Ki8751 (0.75 mg/kg IP), a selective VEGFR2 tyrosine kinase inhibitor. In pups submitted to the NEC protocol, VEGFR2 inhibition abrogated the protective effect of DMOG on villous endothelial cell proliferation (1.6+0.4 vs. VT 11.5+0.88 cells/field, P<0.0001) to a level similar to VEGFR2 inhibition alone (1.6+0.4 vs. 1.7+0.3 cells/field, NS; Figure 4b). In addition, in DF DMOG-treated pups, VEGFR2 inhibition decreased endothelial cell proliferation (3.5+0.4 vs. 26.8+1.8 cells/HPF, P<0.0001) to a level similar to VEGFR2 inhibition alone (3.5+0.4 vs. 5.1+0.9 cells/HPF, NS; Figure 4a–c).
However, VEGFR2 inhibition had no effect on IEC proliferation (2.98+0.27 vs. 3.1+0.24, NS) or on DMOG-improved IEC proliferation (5.1+0.32 vs. 4.9+0.50, NS) in pups exposed to experimental NEC (Figure 5).
Discussion
Despite advances in neonatal intensive care medicine, necrotizing enterocolitis continues to be associated with significant morbidity and high mortality in affected infants (2, 3, 4, 5). Currently, there are no specific therapies available for NEC, with treatment limited to supportive care, bowel rest, parenteral nutrition, and empiric antibiotic therapy (3, 4, 5). Although protective factors have been identified that decrease the incidence of NEC in very low birth weight preterm infants, such as human milk (20, 21) and antenatal betamethasone administration to mothers at risk for preterm delivery (22), innovative targeted therapies are needed to protect against intestinal injury in this vulnerable patient population. Here, we found that treatment with DMOG, a PHD inhibitor that stabilizes HIF-1α and upregulates downstream HIF-1 targets including VEGF, significantly decreased mortality and ameliorated intestinal injury in an experimental mouse model of NEC.
Studies have shown increased HIF-1α expression in the neonatal mouse intestine during experimental NEC in vivo (14). Whether the increase in intestinal HIF-1α expression contributes to intestinal injury in murine NEC or protects against it remains unknown. Our laboratory recently demonstrated that the number of cells expressing VEGF, a downstream target of HIF-1α, is decreased in the intestine of human neonates with NEC (6). In our murine NEC model, we found that endogenous VEGF protein expression and both VEGFR2 protein expression and phosphorylation are decreased before tissue necrosis during NEC development (7). Furthermore, administering SU5416 or Ki8751 to mouse pups to block VEGFR2 signaling before the NEC protocol led to an increases severity of tissue injury and mortality (7). Here, we show that treatment with DMOG, a HIF-1α-stabilizing agent, before NEC induction protects against intestinal injury and increases endothelial cell proliferation in the lamina propia of the intestinal villi in neonatal mice. The effect of DMOG on survival, incidence of severe intestinal injury, and endothelial cell proliferation was dependent on VEGFR2 signaling, as it was completely abrogated by Ki8751, a selective inhibitor of VEGFR2 kinase activity.
DMOG is a nonspecific PHD domain enzyme inhibitor and, accordingly, its effects are not limited to the intestinal microvasculature. As HIF has an important role in IEC physiology (12), we also evaluated whether DMOG treatment affects the intestinal epithelium. We found that DMOG administration before NEC induction attenuated the NEC-induced decrease in IEC proliferation. However, the protective effect of DMOG on IEC proliferation persisted when pups were pretreated with a VEGFR2 inhibitor. DMOG administration before NEC induction no longer conferred a protective benefit against mortality or severity of intestinal injury in pups co-treated with VEGFR2 inhibitor, suggesting that DMOG’s protective effect in NEC is mediated via its impact on intestinal endothelial cells rather than on epithelial cells.
Risk factors for NEC include conditions that predispose the intestine to ischemia, including congenital heart disease, birth asphyxia, maternal preeclampsia with associated placental vascular insufficiency, severe anemia, and alterations in mesenteric blood flow (23, 24, 25). Hypoxia alone has been implicated in NEC development, with clinical studies linking lower target oxygen saturations with an increased incidence of NEC in extremely premature infants (26). Our previous studies suggest that the intestinal microvasculature likely has an important role in NEC pathogenesis (6, 7). Inadequate VEGF–VEGFR2 signaling alters mucosal angiogenesis and contributes to intestinal vascular insufficiency that may predispose premature infants to NEC development when exposed to the stresses of extrauterine life (7). These studies support the notion that stabilizing HIF prevents inadequate intestinal microvasculature development by regulating VEGF–VEGFR2 signaling, thus promoting intestinal perfusion and mesenteric oxygen delivery in premature infants at risk for necrotizing enterocolitis.
The activation of HIFs is part of a wide range of physiological responses that are necessary at the cellular level for tissue survival at low oxygen tension (8). In addition to promoting angiogenesis and tissue regeneration in the setting of ischemia, HIF-1α has a critical role in regulating oxygen homeostasis during fetal and postnatal life (27). A physiological hypoxic environment is required for normal mammalian fetal development, in order to activate HIF-mediated transcriptional regulation of multiple genes with developmentally critical functions (24). When premature infants are exposed to "room air" or ≥21% FiO2, oxygen levels increase to supraphysiologic levels for this developmental stage, which may lead to inactivation of HIF-1α. An untimely decrease in HIF-mediated signaling may contribute to inadequate responses to hypoxic or ischemic insults and can lead to lack of VEGF production during a critical window of intestinal vascular development.
The expression of VEGF and other downstream HIF-1 targets is tightly developmentally regulated in the developing fetus. Nonspecific systemic upregulation of HIF-1 targets such as VEGF at the wrong time could have dangerous unintended effects on several organ systems outside of the gastrointestinal tract (28, 29). In particular, systemically inhibiting PHD in premature infants whose microvasculature is still developing may increase the risk of retinopathy of prematurity, a potentially blinding disease of premature infants caused by abnormal vascularization of the developing retina (28). Furthermore, although numerous studies report that HIF-1 is beneficial for cells exposed to hypoxia or undergoing stress (18, 19), prolonged HIF-1 activation has been linked to loss of gut barrier function, bacterial translocation, and intestinal cell apoptosis, ultimately resulting in villous injury (30). Therefore, before DMOG can be considered as a potential innovative therapy for NEC, additional studies clarifying its mechanism of protection and its effects on other signaling pathways are warranted (15, 31). Careful evaluation of the drug’s effects on other organs and optimal timing for therapeutic intervention will be critical. Treatment strategies that selectively inhibit PHD enzyme activity in the intestine may be preferable.
Systemic DMOG treatment has proven effective in models of vascular injury. Specifically, in a rat model of stroke, increased regional cerebral blood flow, reduced infarct volumes, and improved behavior both 24 h and 8 days after brain injury occurred in rats that were given DMOG before and after transient middle cerebral artery occlusion (18). These protective effects were associated with increased mRNA and protein levels of the HIF-1 targets VEGF and eNOS (18). PHD inhibition has also been shown to be a promising strategy to protect against ischemic injury in several clinical applications (18, 19, 32). Although the role of HIFs in neonatal NEC is not well understood, there is accumulating evidence that hypoxia-induced changes in HIF-mediated genes are important in intestinal diseases characterized by ischemia and inflammation (33, 34). As oxygen-sensing PHDs control transcriptional adaptation to intestinal tissue hypoxia, therapeutic strategies to modulate these hypoxia-signaling pathways for the treatment of inflammatory intestinal diseases have become an important target for study (31). In this study, we demonstrated that PHD inhibition with DMOG drastically improved survival and protected against intestinal injury in an experimental NEC model. Furthermore, our in vivo findings suggest that this may be, at least in part, via improving microvascular integrity as suggested by upregulation of VEGF and increased intestinal villus endothelial cell proliferation.
Currently, there are no targeted therapies for NEC. In this study, to investigate the role of HIFs in NEC pathogenesis, we utilized DMOG, a PHD inhibitor, to stabilize HIF alpha subunits before exposure to the NEC protocol. We found that: (i) PHD inhibition by DMOG before NEC induction significantly decreased mortality and the incidence of severe intestinal injury in neonatal mice subjected to an experimental NEC model whether administered enterally or intraperitonially; (ii) PHD inhibition by DMOG preserved both villous endothelial cell and epithelial cell proliferation during NEC; (iii) PHD inhibition by DMOG increased intestinal VEGF expression in DF pups; (iv) VEGFR2 inhibition by Ki8751, a selective inhibitor of VEGFR2 tyrosine kinase activity, abrogated DMOG’s protective effect on villous endothelial cell proliferation without affecting DMOG’s protective effect on IEC proliferation; and (v) VEGFR2 inhibition abrogated DMOG's protective effect on intestinal injury or mortality. Taken together, these findings provide a basis for testing therapeutic strategies that can selectively inhibit PHD enzyme activity and preserve VEGF–VEGFR2 signaling in neonates at risk for NEC to prevent the development of the disease. Further investigation is needed to evaluate whether specific inhibition of individual PHDs or activation of specific HIF isoforms are effective therapeutic strategies to protect against intestinal injury in NEC.
References
Hopkins GB, Gould VE, Stevenson JK, Oliver TK Jr . Necrotizing enterocolitis in premature infants. A clinical and pathological evaluation of autopsy material. Am J Dis Child 1970;120:229–32.
De Plaen IG . Inflammatory signaling in necrotizing enterocolitis. Clin Perinatol 2013;40:109–24.
Lin PW, Nasr TR, Stoll BJ . Necrotising enterocolitis: recent scientific advances in pathophysiology and prevention. Semin Perinatol 2008;32:70–82.
Lin PW, Stoll BJ . Necrotizing enterocolitis. Lancet 2006;368:1271–83.
Caplan MS. Neonatal necrotizing enterocolitis: clinical observations, pathophysiology, and prevention. In: Martin RJ, Fanaroff AA, Walsh MC, eds. Fanaroff and Martin’s Neonatal-Perinatal Medicine. 9th edn. St Louis, MO: Elsevier, 2011:1431-42.
Sabnis A, Carrasco R, Liu SXL et al. Intestinal vascular endothelial growth factor is decreased in necrotizing enterocolitis. Neonatology 2015;107:191–8.
Yan X, Managlia E, Liu SXL et al. Lack of VEGFR2 signaling causes maldevelopment of the intestinal microvasculature and facilitates necrotizing enterocolitis in neonatal mice. Am J Physiol Gastrointest Liver Physiol 2016;310:G716–25.
Shah YM . The role of hypoxia in intestinal inflammation. Mol Cell Pediatr 2016;3:1.
Lee JW, Bae SH, Jeong JW, Kim SH, Kim KW . Hypoxia-inducible factor (HIF-1)alpha: its protein stability and biological functions. Exp Mol Med 2004;36:1–12.
Avraham-Davidi I, Yona S, Grunewald M et al. On-site education of VEGF-recruited monocytes improves their performance as angiogenic and arteriogenic accessory cells. J Exp Med 2013;210:2611–25.
Grunewald M, Avraham I, Dor Y et al. VEGF-induced adult neovascularization: recruitment, retention, and role of accessory cells. Cell 2006;124:175–89.
Colgan SP, Taylor CT . Hypoxia: an alarm signal during intestinal inflammation. Nat Rev Gastroenterol Hepatol 2010;7:281–7.
Schofield CJ, Ratcliffe PJ . Oxygen sensing by HIF hydroxylases. Nat Rev Mol Cell Biol 2004;5:343–54.
Baregamian N, Rychahou PG, Hawkins HK, Evers BM, Chung DH . Phosphatidylinositol 3-kinase pathway regulates hypoxia-inducible factor-1 to protect from intestinal injury during necrotizing enterocolitis. Surgery 2007;142:295–302.
Jaakkola P, Mole DR, Tian YM et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 2001;292:468–72.
Tian R, Liu SXL, Williams C et al. Characterization of a necrotizing enterocolitis model in newborn mice. Int J Clin Exp Med 2010;3:293–302.
Kubo K, Shimizu T, Ohyama S et al. Novel potent orally active selective VEGFR-2 tyrosine kinase inhibitors: synthesis, structure-activity relationships, and antitumor activities of N-phenyl-N=-{4-(4-quinolyloxy)phenyl} ureas. J Med Chem 2005;48:1359–66.
Nagel S, Papdakis M, Chen R et al. Neuroprotection by dimethyloxalylglycine following permanent and transient focal cerebral ischemia in rats. J Cereb Blood Flow Metab 2011;31:132–43.
Poynter JA, Manukyan MC, Wang Y et al. Systemic pretreatment with dimethyloxalylglycine increases myocardial HIF-1 alpha and VEGF production and improves functional recovery after acute ischemia/reperfusion. Surgery 2011;150:278–83.
Sisk PM, Lovelady CA, Dillard RG, Gruber KJ, O'Shea TM . Early human milk feeding is associated with a lower risk of necrotizing enterocolitis in very low birth weight infants. J Perinatol 2007;27:428–33.
Meinzen-Derr J, Poindexter B, Wrage L et al. Role of human milk in extremely low birth weight infants' risk of necrotizing enterocolitis or death. J Perinatol 2009;29:57–62.
Bauer CR, Morrison JC, Poole WK et al. A decreased incidence of necrotizing enterocolitis after prenatal glucocorticoid therapy. Pediatrics 1984;73:682–8.
Patel RM, Knezevic A, Shenvi N et al. Association of red blood cell transfusion, anemia, and necrotizing enterocolitis in very low-birth-weight infants. JAMA 2016;315:889.
Nowicki PT . Effects of sustained flow reduction on postnatal intestinal circulation. Am J Physiol 1998;275:G758–68.
Nankervis CA, Reber KM, Nowicki PT . Age-dependent changes in the postnatal intestinal microcirculation. Microcirculation 2001;8:377–87.
The BOOST II United Kingdom, Australia, and New Zealand Collaborative Groups. Oxygen saturation and outcomes in preterm infants. N Engl J Med 2013;368:2094–104.
Park AM, Sanders TA, Maltepe E . Hypoxia-inducible factor (HIF) and HIF-stabilization agents in neonatal care. Semin Fetal Neonatal Med 2010;15:196–202.
Gogat K, Le Gat L, Van Den Berghe L et al. VEGF and KDR gene expression during human embryonic and fetal eye development. Invest Ophthalmol Vis Sci 2004;45:7–14.
Gerber HP, Hillan KJ, Ryan AM et al. VEGF is required for growth and survival in neonatal mice. Development 1999;126:1149–59.
Feinman R, Deitch EA, Watkins AC et al. HIF-1 mediates pathogenic inflammatory responses to intestinal ischemia-reperfusion injury. Am J Physiol Gastrointest Liver Physiol 2010;299:G833–43.
Chan MC, Holt-Martyn JP, Schofield CJ et al. Pharmacological targeting of the HIF hydroxylases – a new field in medicine development. Mol Aspects Med 2016;47-48:54–75.
Elzschig H, Bratton DL, Colgan SP . Targeting hypoxia signaling for the treatment of ischaemic and inflammatory diseases. Nat Rev Drug Discov 2014;13:852–69.
Clambey ET, McNamee EN, Westrich JA et al. Hypoxia-inducible factor-1 alpha-dependent induction of FoxP3 drives regulatory T-cell abundance and function during inflammatory hypoxia of the mucosa. Proc Natl Acad Sci USA 2012;109:E2784–93.
Eltzschig HK, Carmeliet P . Hypoxia and inflammation. N Engl J Med 2011;364:656–665.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Statement of Financial Support
This study was supported by funding from Friend of Prentice (FOP) and National Institutes of Health grant R01HD060876 (I.D.P.), RO1DK064240, and RO1GM117628 (X.D.T.). Also, the study was supported in part by the Stanley Manne Children's Research Institute and the Ann & Robert H. Lurie Children's Hospital of Chicago.
Rights and permissions
About this article

Cite this article
Bowker, R., Yan, X., Managlia, E. et al. Dimethyloxalylglycine preserves the intestinal microvasculature and protects against intestinal injury in a neonatal mouse NEC model: role of VEGF signaling. Pediatr Res 83, 545–553 (2018). https://doi.org/10.1038/pr.2017.219
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/pr.2017.219
This article is cited by
-
Recombinant IGF-1/BP3 protects against intestinal injury in a neonatal mouse NEC model
Pediatric Research (2024)
-
Necrotizing enterocolitis: recent advances in treatment with translational potential
Pediatric Surgery International (2023)
-
Advances in our understanding of the molecular pathogenesis of necrotizing enterocolitis
BMC Pediatrics (2022)
-
Macrophage-derived IGF-1 protects the neonatal intestine against necrotizing enterocolitis by promoting microvascular development
Communications Biology (2022)
-
Hope on the horizon: promising novel therapies for necrotizing enterocolitis
Pediatric Research (2020)
