
Abstract
Background
Fetal alcohol syndrome (FAS) is caused by maternal alcohol consumption during pregnancy; although additional factors might be involved, as development and severity are not directly related to alcohol intake. The abnormal glycosylation caused by alcohol might play a role in FAS according to the clinical similarities shared with congenital disorders of glycosylation (CDG). Thus, mutations underlying CDG, affecting genes involved in glycosylation, could also be involved in FAS.
Methods
A panel of 74 genes involved in N-glycosylation was sequenced in 25 FAS patients and 20 controls with prenatal alcohol exposure. Transferrin glycoforms were evaluated by HPLC.
Results
Rare (minor allele frequency<0.009) missense/splice site variants were more frequent in FAS than controls (84% vs. 50%; P=0.034, odds ratio: 5.25, 95% confidence interval: 1.3–20.9). Remarkably, three patients, but no controls, carried variants with functional effects identified in CDG patients. Moreover, the patient with the most severe clinical phenotype was the only one carrying two variants with functional effects. Family studies support that the combination of a genetic defect and alcohol consumption during pregnancy might have a role in FAS development.
Conclusions
Our study supports that the rare variants of genes involved in N-glycosylation could play a role in the development and severity of FAS under prenatal alcohol exposure.
Similar content being viewed by others
Main
Fetal alcohol spectrum disorders (FASDs) include all syndromes resulting from prenatal alcohol exposure (1, 2, 3, 4). The term fetal alcohol syndrome (FAS) is used to name the most severe disorder of this spectrum with different birth defects: cranio-facial abnormalities, growth deficiencies, and central nervous system problems (5). The phenotype depends basically on the timing and the level of alcohol exposure, with the most serious consequences in those fetuses exposed to high concentrations during the initial weeks of pregnancy (6, 7, 8, 9); however, additional factors may be involved as some exposed pregnancies develop FAS, whereas others do not, and the severity of patients is highly heterogeneous. The estimated prevalence of FAS is 1–3/1,000 live births and constitutes the principal cause of non-inherited mental disability (6, 7, 10, 11).
Diverse pathophysiological mechanisms have been proposed to underlie FAS, but none of them completely clarifies this disorder. Recently, a new pathogenetic model suggests that glycosylation defects underlie FAS based on the interference of alcoholism in the glycosylation (12, 13), a major post-translational modification (14), and the overlapping clinical presentations of FAS and congenital disorders of glycosylation (CDG) (15). CDG comprises a broad range of rare and mostly severe genetic diseases with multisystem clinical presentations caused by biallelic mutations in genes involved in glycosylation (16).
A new mechanism of hypoglycosylation has been proposed after the identification of patients with transient disorders of glycosylation: the combination of single genetic defects in genes involved in N-glycosylation and alcohol intake (17). The aim of this study was to evaluate whether this mechanism might also underlie FAS.
Methods
Subjects
The study included 25 patients with FAS meeting the 4-Digit Code (18) and/or the Revised Institute of Medicine criteria for clinical diagnosis (2), retrospectively recruited from the Medical Genetics & Dysmorphology Unit (Hospital Clínico Universitario Virgen de la Arrixaca, Murcia, Spain) and Clinical Genetics Unit (Hospital Vall d´Hebron, Barcelona, Spain). Comprehensive clinical evaluation was performed to exclude other syndromic conditions (phenocopies). Available family members of probands were also studied.
As controls, 20 healthy children with documented prenatal alcohol exposure in utero by fatty acid ethyl ester testing in meconium with ethylglucuronide values between 0.5 and 1.5 nmol/g, but with completely normal development and without any clinical feature of FAS were recruited from Unitat de Recerca Infància I Entorn (Institut Hospital del Mar d'Investigaciones Mèdiques, Barcelona, Spain).
Moreover, 392 healthy Spanish blood donors (41.4±13.1 years, 52% male) and 12 PMM2-CDG patients, the most frequent CDG, were also enrolled in this study. The demographic, clinical, laboratory, and genetic features of these PMM2-CDG patients are described elsewhere (19).
The study was approved by the Hospital Virgen de la Arrixaca Ethic Committee. All patients or their relatives and controls provided written informed consent.
Analysis of Plasma Glycoforms and Antithrombin Activity
SDS-PAGE and western blot analysis of plasma proteins (antithrombin, α1-antitrypsin, and transferrin) were performed as described (20). Plasma transferrin glycoforms were also quantified by HPLC (21, 22). Antithrombin activity, significantly decreased in most N-glycosylation defects (23), was determined by using a chromogenic method (24).
Genetic Analysis
Genomic DNA was purified from peripheral blood; purity was assessed using Nanodrop 2000 (Nanodrop Technologies, Nanodrop, Madrid, Spain) and the concentration level determined by fluorometric methods using Qubit 3.0 (Invitrogen, Madrid, Spain).
In FAS patients, 22q11.2 deletion/duplication was evaluated by multiplex ligation-dependent probe amplification (MLPA) with “high density” 22q11 probemix (P250-B1; MRC-Holland, Amsterdam, the Netherlands) as described (1). Confirmation of 22q11.2 microdeletion was performed by fluorescence in situ hybridization using Vysis LSI TUPLE 1 probe (Abbott Laboratories, Madrid, Spain). Comparative genomic hybridization was performed with Agilent 60K oligo array (Agilent Technologies) using a reference genomic DNA male (Promega, Madrid, Spain). Microarray data were extracted and visualized using Feature Extraction software v10.1 and Genomic Workbench software v6.5 (Agilent Technologies, Madrid, Spain).
The protein-coding exons and intron–exon boundaries of 74 genes involved in the N-glycosylation (Supplementary Table S1 online) were sequenced by DNA target capture followed by Ion PGM sequencing. Torrent Suite v5.0.2 software (Madrid, Spain) was used for analysis. Genetic variants were identified using Variant Caller 5.0.2.1 software (Madrid, Spain) using default settings (germline & low stringency). VCF file for each sample was transferred and variants were annotated using Ion Reporter 5.0 (Madrid, Spain).
The association with FAS of variants and the odds ratio (OR) calculation was assessed by individual-variant testing carried out with Fisher’s exact probability test. The number of variants in controls and patients were compared by Student’s t-test, considering statistical significance P<0.05.
Results
The clinical and demographic characteristics of patients with FAS (n=25) are shown in Table 1.
Sequence metrics were similar among patients and controls (Supplementary Table S2). The number of single-nucleotide variations (SNVs) in patients and controls was nearly similar (163.1±21.8 and 171.5±3.5, respectively). Common variants (with minor allele frequency (MAF)>0.09 according to data of the 5000 exomes -https://evs.gs.washington.edu/EVS/-) constituted the main source of genetic variation in patients (90%) and controls (96%). However, the number of rare variants (MAF<0.009) per subject was higher in patients than in controls (1.92±1.4 vs. 0.65±0.67, respectively, P=0.001).
The search for potential pathological variants was initially restricted to those mutations with functional consequences on enzymes involved in N-glycosylation that were identified in CDG patients. Interestingly, three FAS patients carried four mutations affecting PMM2 and ALG6 (Table 2) (25, 26, 27, 28). Control 13 carried a mutation previously described in one CDG patient, but with unknown functional effect (Table 2) (29).
Then we searched for rare variants affecting any of the 19 genes already involved in CDG with potential deleterious consequences according to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar). Fifteen FAS patients and three controls presented SNVs with these features (OR=8.5, 95% confidence interval (CI): 2.3–36.8; P=0.002) (Table 3). Noteworthy, four rare variants (ALG2 p.Gly6Ala, MPI p.Thr350Met, PGM1 rs77043134, and ALG8 p.Arg268Gln) were recurrently identified in two different unrelated patients, but were not found in any controls (Table 3).
Finally, when the analysis was performed with rare SNVs affecting genes not previously associated with CDG but involved in N-glycosylation, 24 variants in 14 FAS patients and 9 variants in 7 controls were identified (Table 4).
All SNVs identified in this study were in heterozygosis state, except for p.Ala28Pro in DPM3 carried by Control 6, which argued against any potential functional consequence for this SNV.
Taken together, 21 FAS patients (84%) as compared to only 10 controls (50%) carried rare SNVs affecting genes potentially involved in N-glycosylation (OR: 5.25, 95% CI: 1.3–20.9; P=0.034) (Table 1). Interestingly, the four patients without rare variants in genes related to the N-glycosylation had only suspicion of prenatal alcohol exposition (Table 1).
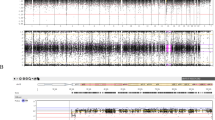
Especially interesting was FAS-1, a patient carrying two functional variants identified previously in CDG patients. The PMM2 p.Phe207Ser variant strongly impairs the phosphomannomutase activity(27) and ALG6 p.Tyr131His also has an important functional effect on the alpha-1,3-glucosyltransferase enzymatic activity (28). In addition, FAS-1 also carried three other rare SNVs affecting genes involved in N-glycosylation: DOLK p.Tyr360Cys, RPN1 p.His238Tyr, and PMM1 p.Ala5Ser with unknown functional consequences (Table 1). The diagnosis of CDG was ruled out in FAS-1 by his normal antithrombin activity (98%) and HPLC transferrin glycoform profile (Figure 1a). Interestingly, FAS-1 has the most severe clinical phenotype in our cohort. The child was born at 33 weeks of gestation by cesarean-section because of abruptio placenta. He had a birth weight of 1,620 g (−1.19 standard deviation score (SDS)), length of 37 cm (−3.89 SDS), and orbital frontal cortex (OFC) of 30 cm (−0.08 SDS). APGAR score was 5/8. At birth, dysmorphic features (including posterior cleft palate) and punctate epiphyseal calcifications were detected. Failure to thrive was present, and he required nasogastric tube feeding until 17 months of age. His psychomotor development was disabled. He sat up at 20 months of age, walked independently at 4 years, and language was retarded. He showed low school performance due to his intellectual disability (IQ: 57) and hyperactivity. He had transient conductive hypoacusia treated by grommets insertion in infancy. Cardiac evaluation by echocardiography was normal. Ophthalmological evaluation showed blepharophimosis and myopia. CT scan showed corpus callosum dysgenesis and dysmorphic ventricles. Skeletal survey in adolescence did not show any calcification; hypoplasia of the 2nd phalange of the 3rd and 5th fingers was observed. Conventional karyotype was normal (46, XY). 22q11.2 microdeletion was ruled out, and a 60K CGH array did not identify any pathogenic copy number variation. Hematology, routine biochemistry, blood clotting analysis, red blood cell plasmalogens, and plasma sterol levels were normal. The patient is currently 18 years old. His weight is 35.5 kg (−3.1 SDS), height is 146 cm (−4.98 SDS), and OFC 49.5 cm (−4.47 SDS). He has microcephaly, blepharophimosis, broad nasal root with flattened nose, long philtrum, thin upper lip, dysplastic and low-set auricles, dental malocclusion, repaired posterior cleft palate, bilateral fifth finger clinodactyly, and third finger camptodactyly (Figure 1b).

Clinical, genetic, and laboratory data about FAS-1 patient and his relatives. (a) HLPC analysis of transferrin glycoforms in a control, a PMM2-CDG patient and FAS-1 patient. (b) Picture of FAS-1 patient. Note his microcephaly, wide eyebrows, blepharophimosis, broad nasal root with flattened nose, long philtrum, thin upper lip, and low-set ears. (c) Family tree of FAS-1 patient. Dark and gray filled symbol represents heterozygous carriers of the PMM2 and ALG6 mutations identified in the proband. Proband is indicated by an arrow.
Family studies in relatives of FAS-1 rendered very interesting results (Figure 1c). The mother, who is now fully rehabilitated, started drinking alcohol after having been fired from work when she was 29 years old, and according to her declaration, consumed high proportions of alcohol (80−100 g/day) during the last three pregnancies. Two of these children, the proband and one brother developed FAS (Figure 1c). Genetic analysis in available samples revealed the PMM2 p.Phe207Ser variant in three siblings and the ALG6 p.Tyr131His in the mother and one sister. Interestingly, the sister with both functional variants did not develop FAS as her embryonic development had not been exposed to alcohol exposition (Figure 1c). Of note, two additional pregnancies under alcohol addiction of the mother rendered one additional FAS, but also a healthy girl (Figure 1c). Unfortunately, these subjects were not available for genetic studies.
Discussion
The broad and multisystem dysfunction caused by defective N-glycosylation genes explains why CDG patients are usually diagnosed in infancy and have severe clinical manifestations (16). On the other hand, diagnosis of CDG is relatively simple. The identification of increased levels of hypoglycosylated forms of multiple proteins (the golden standard is transferrin) allows a secure diagnosis, to be confirmed by sequencing candidate genes (30). The absence of these hypoglycosylated proteins has been the main reason to exclude FAS as another CDG, although both disorders share clinical features. However, the perspective in the field of CDG has noticeably changed after the identification of CDG-like patients, adults without psychomotor disability, heterozygous for an N-glycosylation defect, and showing transient hypoglycosylation (17). The complete deficiency of PMM2 has deleterious consequences usually causing embryonic lethality (31). Biallelic mutations in N-glycosylation genes associated with residual functional activity underlie classical CDG (16). However, a single functional mutation of this pathway is not enough to impair the correct glycosylation of proteins, as demonstrated in CDG parents (20). Why did CDG-like patients carrying only one mutation have transitory hypoglycosylation? We hypothesized that an acquired factor also affecting the same pathway might exacerbate the genetic defect. Alcohol consumption is an excellent candidate not only because it was shared by all CDG-like patients with transitory hypoglycosylation identified in our study (17), but also because there is strong evidence for a deleterious defect of alcohol in N-glycosylation (13). We, therefore, proposed that the provisions of N-glycan precursors required to fully glycosylate N-glycoproteins are insufficient in carriers of a single functional mutation under alcohol exposition, hence leading to hypoglycosylation. We speculated that this mechanism might be involved in FAS. The higher frequency of rare variants, particularly those with well-known functional consequences, of genes involved in N-glycosylation identified in FAS patients compared with controls, also exposed to alcohol, supports our hypothesis and suggests that this combination, through the potentially associated hypoglycosylation, might contribute to the disease. Moreover, our study has identified other genetic variants in genes involved in the N-glycosylation pathway that according to our hypothesis might have functional effects, which must be validated by further studies. This study supplies new evidences on the relevance of genetic-environmental interactions on the development of different disorders. Different environmental factors may trigger the pathogenic consequences of genetic variants for many proteins. Temperature sensitive and conformational mutations are excellent examples (32). We suggest here a new mechanism of genetic-environmental interaction in determinism of FAS, as the defect caused by the genetic variation might reach pathogenic consequences by the synergistic effect of the environmental factor (alcohol).
Our study also explains why CDG-like patients showed no typical CDG clinical manifestations. We propose that a functional mutation affecting N-glycosylation, combined with exposition to alcohol, will cause hypoglycosylation similar to that caused by two mutations in genes involved in N-glycosylation in classical CDG. Once the baby is born, the exposure to alcohol and, thus, the hypoglycosylation also disappears, but not the clinical consequences of the hypoglycosylation during embryogenesis. In contrast, the embryonic development of adults with CDG-like disorders is completely normal because the impaired glycosylation only happens during adulthood, when exposed to alcohol.
In addition, although our study is small and must be validated in larger studies, our data also suggest that the accumulation of functional variants affecting different N-glycosylation genes might modulate the strength of the glycosylation defect, and hence the severity of the FAS phenotype. The best example is patient 1, with two functional variants in different genes. The fact that none of the proteins analyzed (antithrombin, alpha1-antitrypsin, or transferrin) have hypoglycosylated forms (Figure 1) supports that the combination of these two genetic defects by themselves does not cause glycosylation defects, probably because they affect different genes. According to our hypothesis, the combination with alcohol might cross the threshold to cause glycosylation defects. In this particular patient, the alcohol susceptibility might be higher than in the others just because of the two functional variants. In fact, this patient is the one with the most severe clinical picture as it is described in Table 1. Nevertheless, it could be interesting to analyze LLO and/or cell glycosylation in more detail in this patient.
Our study supports the hypothesis that rare variants affecting N-glycosylation genes might increase the risk of FAS when the mother consumed alcohol during pregnancy. Further studies are required to substantiate this hypothesis. It is essential to demonstrate the functional consequences of variants identified in our study, and the hypoglycosylation that might happen in fetuses with one functional variant when exposed to alcohol. Moreover, it is necessary to clarify whether the magnitude of the hypoglycosylation, which may be related to the dose of alcohol or the time exposed to alcohol and the functional consequences of the genetic profile of this pathway, could define the severity of the clinical manifestations in FAS patients.
Finally, the identification of functional variants in genes involved in the N-glycosylation pathway has been restricted to the analysis of known CDG patients. Our study may also assist to identify new functional variants in genes involved in the N-glycosylation pathway. The four rare variants with unknown functional effect identified recurrently in FAS patients, but not in controls are ideal candidates to be explored in further studies.
References
Jalali GR, Vorstman JA, Errami A et al. Detailed analysis of 22q11.2 with a high density MLPA probe set. Hum Mutat 2008;29:433–440.
Hoyme HE, May PA, Kalberg WO et al. A practical clinical approach to diagnosis of fetal alcohol spectrum disorders: clarification of the 1996 institute of medicine criteria. Pediatrics 2005;115:39–47.
Hoyme HE, Kalberg WO, Elliott AJ et al. Updated clinical guidelines for diagnosing fetal alcohol spectrum disorders. Pediatrics 2016;138:e20154256.
Del Campo M, Jones KL . A review of the physical features of the fetal alcohol spectrum disorders. Eur J Med Genet 2016;60:55–64.
Jones KL, Smith DW . Recognition of the fetal alcohol syndrome in early infancy. Lancet 1973;302:999–1001.
Chudley AE, Conry J, Cook JL et al. Fetal alcohol spectrum disorder: Canadian guidelines for diagnosis. CMAJ 2005;172:S1–S21.
Guizzetti M, Costa LG . Cholesterol homeostasis in the developing brain: a possible new target for ethanol. Hum Exp Toxicol 2007;26:355–360.
Kot-Leibovich H, Fainsod A . Ethanol induces embryonic malformations by competing for retinaldehyde dehydrogenase activity during vertebrate gastrulation. Dis Model Mech 2009;2:295–305.
Lipinski RJ, Godin EA, O'leary-Moore SK et al. Genesis of teratogen-induced holoprosencephaly in mice. Am J Med Genet C Semin Med Genet 2010;154C:29–42.
Abdelrahman A, Conn R . Eye abnormalities in fetal alcohol syndrome. Ulster Med J 2009;78:164–165.
van Balkom ID, Gunning WB, Hennekam RC . Fetal alcohol syndrome: an unrecognized cause of intellectual handicap and problem behavior in The Netherlands. Ned Tijdschr Geneeskd 1996;140:592–595.
Waszkiewicz N, Szajda SD, Zalewska A et al. Alcohol abuse and glycoconjugate metabolism. Folia Histochem Cytobiol 2012;50:1–11.
Welti M, Hulsmeier AJ . Ethanol-induced impairment in the biosynthesis of N-linked glycosylation. J Cell Biochem 2014;115:754–762.
Moremen KW, Tiemeyer M, Nairn AV . Vertebrate protein glycosylation: diversity, synthesis and function. Nat Rev Mol Cell Biol 2012;13:448–462.
Binkhorst M, Wortmann SB, Funke S et al. Glycosylation defects underlying fetal alcohol spectrum disorder: a novel pathogenetic model. "When the wine goes in, strange things come out" - S.T. Coleridge, The Piccolomini. J Inherit Metab Dis 2012;35:399–405.
Jaeken J . Congenital disorders of glycosylation. Ann N Y Acad Sci 2010;1214:190–198.
de la Morena-Barrio ME, Martinez-Martinez I, de Cos C et al. Hypoglycosylation is a common finding in antithrombin deficiency in the absenceof a SERPINC1 gene defect. J Thromb Haemost 2016: 1549–1560.
Astley SJ, Clarren SK . Diagnosing the full spectrum of fetal alcohol-exposed individuals: introducing the 4-digit diagnostic code. Alcohol Alcohol 2000;35:400–410.
de la Morena-Barrio ME, Hernandez-Caselles T, Corral J et al. GPI-anchor and GPI-anchored protein expression in PMM2-CDG patients. Orphanet J Rare Dis 2013;8:170.
de la Morena-Barrio ME, Sevivas TS, Martinez-Martinez I et al. Congenital disorder of glycosylation (PMM2-CDG) in a patient with antithrombin deficiency and severe thrombophilia. J Thromb Haemost 2012;10:2625–2627.
del Castillo Busto ME, Meija J, Montes-Bayon M et al. Diophantine analysis complements electrospray-Q-TOF data for structure elucidation of transferrin glycoforms used for clinical diagnosis in human serum and cerebrospinal fluid. Proteomics 2009;9:1109–1113.
Helander A, Husa A, Jeppsson JO . Improved HPLC method for carbohydrate-deficient transferrin in serum. Clin Chem 2003;49:1881–1890.
Linssen M, Mohamed M, Wevers RA et al. Thrombotic complications in patients with PMM2-CDG. Mol Genet Metab 2013;109:107–111.
Hernandez-Espinosa D, Minano A, Martinez C et al. L-asparaginase-induced antithrombin type I deficiency: implications for conformational diseases. Am J Pathol 2006;169:142–153.
Bjursell C, Erlandson A, Nordling M et al. PMM2 mutation spectrum, including 10 novel mutations, in a large CDG type 1A family material with a focus on Scandinavian families. Hum Mutat 2000;16:395–400.
Matthijs G, Schollen E, Van Schaftingen E et al. Lack of homozygotes for the most frequent disease allele in carbohydrate-deficient glycoprotein syndrome type 1A. Am J Hum Genet 1998;62:542–550.
Vega AI, Perez-Cerda C, Abia D et al. Expression analysis revealing destabilizing mutations in phosphomannomutase 2 deficiency (PMM2-CDG): expression analysis of PMM2-CDG mutations. J Inherit Metab Dis 2011;34:929–939.
Westphal V, Xiao M, Kwok PY et al. Identification of a frequent variant in ALG6, the cause of congenital disorder of glycosylation-Ic. Hum Mutat 2003;22:420–421.
Vleugels W, Schollen E, Foulquier F et al. Screening for OST deficiencies in unsolved CDG-I patients. Biochem Biophys Res Commun 2009;390:769–774.
Quintana E, Navarro-Sastre A, Hernandez-Perez JM et al. Screening for congenital disorders of glycosylation (CDG): transferrin HPLC versus isoelectric focusing (IEF). Clin Biochem 2009;42:408–415.
Thiel C, Lubke T, Matthijs G et al. Targeted disruption of the mouse phosphomannomutase 2 gene causes early embryonic lethality. Mol Cell Biol 2006;26:5615–5620.
de la Morena-Barrio ME, Sandoval E, Llamas P et al. High levels of latent antithrombin in plasma from patients with antithrombin deficiency. Thromb Haemost 2017;117:880–888.
Acknowledgements
We acknowledge Irene Martínez-Martínez and Salvador Espín for their assistance on HPLC analysis.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
AUTHOR CONTRIBUTIONS
M.E.M.-B., J.C., E.G.-N., and V.V. designed research, analyzed the data, and wrote the paper. M.J.B.-M., V.L.-G., L.M.-R., O.G.-A., M.D.C., and E.G.-N. collected patients and clinical data and revised the paper. R.L.-G., M.E.M.-B., and A.M. performed biochemical and genetics experiments. A.I.M. and J.P. performed NGS analysis and genetic analysis.
STATEMENT OF FINANCIAL SUPPORT
MEM-B holds a postdoctoral fellowship from Fundacion Espanola de Trombosis y Hemostasia, Spain. This work was supported by PI15/00079 and CB15/00055 from ISCIII & FEDER; 19873/GERM/15 Fundacion Seneca.
Supplementary material is linked to the online version of the paper at
Supplementary information
Rights and permissions
About this article

Cite this article
de la Morena-Barrio, M., Ballesta-Martínez, M., López-Gálvez, R. et al. Genetic predisposition to fetal alcohol syndrome: association with congenital disorders of N-glycosylation. Pediatr Res 83, 119–127 (2018). https://doi.org/10.1038/pr.2017.201
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/pr.2017.201
