
Abstract
Background:
Disturbances in cerebral blood flow (CBF) and brain oxygenation (PbO2) are present early after pediatric cardiac arrest (CA). CBF-targeted therapies improved neurological outcome in our CA model. To assess the therapeutic window for CBF- and PbO2-targeted therapies, we propose to determine if CBF and PbO2 disturbances persist at 24 h after experimental pediatric CA.
Methods:
Regional CBF and PbO2 were measured at 24 h after asphyxial CA in immature rats (n = 26, 6–8/group) using arterial spin label MRI and tissue electrodes, respectively.
Results:
In all regions but the thalamus, CBF recovered to sham values by 24 h; thalamic CBF was >32% higher after CA vs. sham. PbO2 values at 24 h after CA in the cortex and thalamus were similar to shams in rats who received supplemental oxygen, however, on room air, cortical PbO2 was lower after CA vs. shams.
Conclusion:
CBF remains increased in the thalamus at 24 h after CA and PbO2 is decreased to hypoxic levels in cortex at 24 h after CA in rats who do not receive supplemental oxygen. Given the enduring disturbances in this model and the lack of routine CBF or PbO2 monitoring in patients, our data suggest the need for clinical correlation.
Similar content being viewed by others

Main
Hypoxic ischemic encephalopathy is the prime limiting factor for intact neurological survival after pediatric cardiac arrest (CA). Among children with sustained return of spontaneous circulation after CA, 76% of children in the out-of-hospital setting and 53% of children resuscitated after in-hospital CA develop neurological impairment (1). Cerebral blood flow (CBF) disturbances and cerebral hypoxia might contribute to a secondary insult after CA.
In a pediatric asphyxial CA model of 9 min in postnatal day 16–18 rats, CBF disturbances are region-specific. Cortical blood flow is decreased vs. baseline from 10 to 150 min post-CA, whereas thalamic blood flow is increased vs. baseline from 5 to 30 min post-CA, followed by return to baseline values from 60 to 180 min (2). Brain tissue oxygen tension (PbO2) follows a similar pattern of cortical hypoxia from 30 to 120 min and early thalamic hyperoxia at 5–10 min, followed by return to baseline levels by 120 min (3). Supplemental oxygen increased thalamic and cortical PbO2 at 120 min after CA. Mitigation of CBF disturbances immediately after resuscitation improved neurological outcome after pediatric asphyxial CA: administration of albumin increased cortical blood flow, while administration of the antioxidant nitroxide polynitroxyl albumin (PNA) decreased thalamic hyperemia, and each improved long-term functional outcome assessed by the Morris water maze performance in our model (4).
To delineate the scope, magnitude, and duration of time window for targeted CBF and PbO2 promoting strategies in our model, our objective was to determine if CBF and PbO2 disturbances are present at 24 h after experimental CA in PND 17 rats. We hypothesized that the regional CBF and PbO2 disturbances seen early after CA resolve by 24 h after CA and that supplemental oxygen would increase regional PbO2 post-CA. We used arterial spin labeling (ASL) magnetic resonance imaging (MRI) assessment to quantify regional CBF and measured PbO2 in cortex and thalamus across a spectrum of FiO2 levels. Our results indicate that there are enduring CBF disturbances in thalamus and brain tissue hypoxia in the cortex at 24 h after CA in our pediatric CA model.
Results
Physiological data from the groups of rats that underwent sham surgery or CA are presented in Table 1 . MAP in rats who underwent PbO2 measurement was lower at 24 h after CA compared with shams (52 ± 1 vs. 64 ± 3 mmHg, P < 0.05). The other variables did not differ after CA compared with shams. Due to the small size of the rats, we had to limit the repeated sampling of blood post-CA, and thus for the PbO2 CA group at FiO2 = 0.21, the pH, PaCO2, and PaO2 values are computed from two rats. The rest of the values in the table are computed from four to six rats.
Regional CBF at 24 h After CA: Thalamic CBF Is Increased Compared With Shams
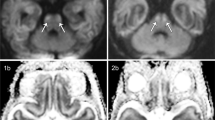
CBF maps from a sham-operated rat and a rat imaged at 24 h after CA are represented in Figure 1a . Figure 1b represents regional CBF data at 24 h after sham surgery or CA. At 24 h after CA, cortical CBF was comparable with shams. Likewise, hippocampal and amygdalic CBF were comparable with shams at 24 h after CA ( Figure 1b ). In contrast, thalamic CBF was > 32% higher after CA compared with sham (283 ± 21 vs. 213 ± 6 ml/100g/min, CA vs. sham, respectively, P < 0.05, Figure 1b ).

Regional cerebral blood flow at 24 h after cardiac arrest. (a) T2-weighted image (left) showing the regions of interest used for the CBF study, yellow is cortex, red is hippocampus, green is thalamus, and blue is the region of the amygdala. Representative regional CBF maps in a sham-operated rat (middle) and a rat imaged at 24 h after CA (right). Increased CBF in the thalamic area is present at 24 h after CA vs. sham. (b) Regional CBF data at 24 h after sham surgery (solid boxes) or CA (dashed boxes). The box plots display the median ± one SD, and maximum and minimum values. Rats received FiO2 = 0.5. (*P < 0.01 vs. sham) (n = 6/group).
PbO2 Levels at 24 h After CA: Cortical PbO2 Are Decreased to Hypoxic Levels in Room Air at 24 h After CA
When rats received FiO2 = 0.5 or 1.0 at 24 h post-CA, PbO2 in the cortex and thalamus were above what is generally considered to be the ischemic threshold (10 mm Hg) (5,6), and comparable to shams ( Figure 2 ). Cortical PbO2 was 61 ± 9 mm Hg in shams and 47 ± 15 mm Hg after CA for FiO2 = 0.5 (P = 0.4), and 54 ± 11 mm Hg in shams and 42 ± 14 mm Hg after CA for FiO2 = 1.0 (P = 0.5, Figure 2a ). Similarly, in the thalamus, PbO2 was 68 ± 8 mm Hg in shams and 77 ± 15 mm Hg after CA for FiO2 = 0.5 (P = 0.6), and, 92 ± 19 mm Hg in shams and 96 ± 33 mm Hg after CA for FiO2 = 1.0 (P = 0.5, Figure 2b ).

Cortical and thalamic brain tissue oxygen tension at 24 h after cardiac arrest. Cortical (a) and thalamic (b) PbO2 for sham surgery (solid boxes) and CA animals (dashed boxes), at 24 h after CA while the rats were administered different oxygen concentrations: 50% oxygen (FIO2 = 0.5), 100% oxygen (FiO2 = 1), and room air (FiO2 = 0.21). The box plots display the median ± one standard deviation, and maximum and minimum values. Cortical and thalamic hypoxia is present when the rats inhale FiO2 = 0.21 (*P < 0.016 vs. sham) (n = 6–8/group).
However, when the rats received room air (FiO2 = 0.21), cortical PbO2 levels were 13.1 ± 3.2 mm Hg in shams and markedly and significantly reduced to a value of 2.6 ± 1.6 mm Hg at 24 h after CA (P < 0.016 shams vs. CA, Figure 2a ). Thalamic PbO2 levels while rats received room air were 19.2 ± 3.1 mm Hg in shams but 9.1 ± 2.2 mmHg at 24 h after CA, however this did not reach significance when corrected for multiple comparisons (P = 0.02 shams vs. CA, Figure 2b ). PaO2 values for shams and after CA at FiO2 = 0.21 were similar: 55 ± 5 and 49 ± 0.1 mm Hg, respectively ( Table 1 ).
Discussion
Remarkably, to our knowledge, this is the first report of regional CBF and PbO2 at 24 h after the insult in a preclinical model of pediatric asphyxial CA. Our data show that cortical blood flow, previously observed to be critically reduced at 3 h after CA, returns to normal values at 24 h after CA. Conversely, CBF in the thalamus is increased at 24 h after CA. PbO2 levels return to normal in the cortex and thalamus while rats receive supplemental oxygen. However, cortical PbO2 levels are below the ischemic threshold while rats receive room air-despite similar arterial PaO2 values on room air in sham and post-CA rats at 24 h.
All regions except for the thalamus regain baseline CBF values. In the context of our prior studies showing improved CBF and behavioral recovery with CBF promoting therapies, the current results suggest that strategies to mitigate cortical hypoperfusion are likely to show maximal efficacy if started early after CA. Consistent with this finding, CBF promoting therapies were effective in our model when started immediately after resuscitation (7,8). However, we cannot completely rule out potential efficacy of CBF promoting strategies at 24 h after CA in our model because either augmentation of CBF or raising FiO2 could potentially mitigate the tissue hypoxia seen in the cortex at 24 h when the rats received room air.
We observed that thalamic CBF is increased at 24 h after CA. The thalamus represents one of the vulnerable regions post-CA in humans (9) and animal models (10,11,12). Functional consequences of thalamic injury include deficits in attention (13,14), sleep wake cycle (15), transmission of information to the cerebral cortex (16), and movement disorders, pathologies present in survivors of CA (17). In our pediatric asphyxial CA model, there is evidence of disruption of thalamic circuit function after CA (18). Single neuron recordings of thalamocortical neurons after CA demonstrated increased firing rates at 48–72 h after CA. These pathophysiologic disturbances were associated with histologic evidence of injury in the thalamic reticular nucleus and ventral posterior medial nucleus, including extensive degeneration of neurites in the somatosensory nucleus and activation of microglia in the reticular nucleus (18,19).
High CBF in the thalamus in the context of normal PbO2 may reflect neurovascular coupling at 24 h after the insult and is consistent with the increased thalamic firing rates in our model after CA. However, our data also suggest that despite the increase in CBF, the thalamus may remain vulnerable to tissue hypoxia at 24 h given what would be considered critical reductions in PbO2 to levels < 10 mmHg on room air in some rats ( Figure 2b ). Indeed, some have suggested that even higher levels of PbO2 should be targeted after brain injury (5). It is also unclear as to whether or not the delayed increase in CBF in thalamus is beneficial or detrimental. We previously reported that administration of the antioxidant nitroxide PNA attenuated the early increase in thalamic CBF post CA in our model and improved behavioral outcomes (4). However, whether this same mechanism underlies the delayed increases in thalamic CBF remain to be defined.
In addition to the findings in the thalamus, cortical PbO2 levels at 24 h were reduced in our model at what would be generally accepted to represent a critical values in rats receiving room air ( Figure 2a ) (5,6,20). Supplemental oxygen improved PbO2 in both cortex and thalamus by 24 h after CA ( Figure 2 ). These data combined with our previous observations of cortical hypoxia early after CA suggest that monitoring of PbO2 could be essential to identifying cortical hypoxia and potentially guiding therapy, even at time points as late as 24 h after CA (3). Importantly, although hypoxic ischemic encephalopathy represents the preeminent morbidity after CA, cerebral monitoring is not employed after CA. Recent studies in adults after CA suggest that ICP monitoring coupled to cerebral microdialysis may be important to guiding brain-directed therapy, and thus PbO2 monitoring after CA might be feasible (21). Moreover, a noninvasive approach to the assessment of cortical oxygenation, near infrared spectroscopy (NIRS), represents a viable cerebral monitoring modality (22,23,24), however it is not standard of care in clinical management of pediatric CA and deserves to be systematically studied post-CA.
In our experiments, rats who sustained CA as well as shams who received room air while being mechanically ventilated had low values of PaO2, likely secondary to dependent atelectasis (3,25). Remarkably, however, only rats who sustained CA had critically low cortical PbO2 values, despite similar low values of PaO2 for both shams and rats who sustained CA. Our experiments reveal a vulnerability of the post-hypoxic brain to moderate hypoxemia and suggest that even at delayed time points of 24 h post-CA the cortex is unable to compensate for hypoxemia, possibly secondary to loss of the ability to vasodilate in response to hypoxemic conditions. These results suggest that even modest levels of hypoxemia should be avoided after cardiac arrest.
We observed blood pressure differences between sham rats ventilated with room air or supplemented with oxygen. This may be related to the vasoconstrictor effect of oxygen on peripheral arteries. The MAP values are well within the autoregulatory range of PND 17 rats, and are unlikely to have influenced the PbO2 values (26,27).
In conclusion, enduring disturbances of CBF and PbO2 are detectable at 24 h after asphyxial CA in the developing rat brain. Thalamic CBF is increased at 24 h after CA while PbO2 reaches critical hypoxic values in cortex and has a trend towards being reduced in thalamus at 24 h after CA in rats without oxygen supplementation. Given the enduring disturbances in this model, and the lack of routine CBF or PbO2 monitoring in patients, our data suggest the need for clinical correlation. Also, given the benefit of prior reports of strategies to normalize immediate post-resuscitation CBF and PbO2 in our model, assessment of the therapeutic window is warranted.
Methods
We used postnatal day 16–18 male Sprague-Dawley rats (30–45 g), n = 26, 6–8 rats/group. Studies were approved by the University of Pittsburgh animal care committee. One group of rats was used for quantification of regional CBF, and another for measurement of cortical and thalamic PbO2. Sham-operated rats served as controls for CBF and PbO2 measurement.
Asphyxial Insult
We used an established model of asphyxial CA developed in our laboratory (28). Rats were anesthetized with 3% isoflurane/50% N2O/balance oxygen until unconscious. Trachea was intubated with an 18-gauge angiocatheter and mechanical ventilation was initiated. Central femoral arterial and venous catheters were inserted via left inguinal cut-down. Anesthesia was maintained with 1% isoflurane/50% N2O/O2. A bolus of vecuronium (1 mg/kg, iv) was administered 2 min prior to asphyxia to prevent respirations. Asphyxia was produced by disconnecting the tracheal tube from the ventilator for 9 min. Rats were resuscitated by reconnecting the ventilator, iv administration of epinephrine 0.005 mg/kg and sodium bicarbonate 1 mEq/kg, and manual chest compressions until return of spontaneous circulation. One hour after return of spontaneous circulation, trials of weaning the ventilator were started. If spontaneous respiration did not occur within 30 s after the start of the trial, the tracheal tube was reconnected to the ventilator and another trial was performed in 15 min. In our model, the first weaning trial is successful at 1 h after resuscitation in 85% of the rats. The rats were returned to their cages until 24 h post-resuscitation. At 24 h post-resuscitation, CBF or PbO2 was measured in separate groups of rats. For the 24 h CBF and PbO2 measurements the rats underwent tracheal intubation and arterial and venous catheter placement as described above except that the catheters were inserted via right inguinal cut-down. Isoflurane was then discontinued to mimic clinical care and intravenous analgesia and neuromuscular blockade were started using a fentanyl infusion at 50 μg kg−1 h−1 and vecuronium infusion at 5 mg kg−1 h−1. A 30 min period of isoflurane washout was performed before the CBF and PbO2 measurements were taken to reduce the confounding effects of inhaled anesthetics on CBF and PbO2 (29). The rats were ventilated with 50% O2. Sham rats underwent identical procedures, without CA or resuscitation medications.
Cerebral Blood Flow Assessment
Regional CBF was measured using the method of ASL-MRI (n = 6 rats/group). Data acquisition and data analysis using ASL-MRI were reported by our laboratory previously (2). MR studies were performed on a 7-Tesla, 21-cm-bore Bruker Biospec system equipped with a 12-cm-diameter shielded gradient insert and a 72 mm volume RF coil. Data were acquired for T2-weighted images, perfusion images (30), spin-lattice relaxation time maps of tissue water (T1obs) (31), and spin-labeling efficiency measurements within the carotid arteries (32). T2-weighted spin-echo images were acquired with the following parameters: field of view (FOV) = 3 cm, slice thickness = 1 mm, interslice distance = 2 mm, TR/TE = 2,500/40 ms, NA=2, five slices, and matrix = 128 × 70. Perfusion images were acquired using ASL (30) with continuous flow-induced adiabatic inversion for 2 s with a constant-amplitude RF pulse in the presence of a 1 G/cm field gradient along the head-foot axis of the animal. The labeling pulse for the inversion plane was positioned ± 2 cm from the perfusion detection plane. The parameters were: matrix = 64 × 40, zero-filled to 64 × 64; TR = 2,000 ms; summation of three echoes; TE = 10, 20, and 30 ms; and NA = 2. Spin-labeling efficiency was determined from intensities within the carotid arteries with the use of gradient-echo images (32). Images were acquired 1 cm posterior to the plane selected for perfusion detection, with spin-labeling applied at ± 10 mm. The perfusion encoding parameters were the same as those mentioned above, and included a 45° flip angle, eight averages, TR/TE = 100/9.6 ms, and matrix = 256 × 256. The spin-lattice relaxation time of tissue water (T1obs) (31) was measured from a series of spin-echo images acquired with variable TR (TR = 8,000, 4,300, 2,300, 1,200, 650, 350, 185, and 100 ms). Other parameters are as follows: TE = 9 ms, two averages, and matrix = 64 × 40. Image Analysis: All image processing was performed with the ParaVision 5.0 image analysis software (Bruker Biospin, Billerica, MA). Regions of interest were defined in the left and right hemispheres guided by assignments from a rat brain atlas and included the cortex, hippocampus, thalamus, and amygdala/piriform cortex. Pixel-by-pixel maps of (MC - ML) • MC were generated from the perfusion data (where MC is the magnetization intensity from the control image, and ML is the magnetization intensity from the labeled image). T1obs maps were generated from the series of variable TR images by a three-parameter nonlinear fit to, M(TR) = M0 (1 – A exp(-TR/T1obs)), where M(TR) is the signal intensity for each TR value, M0 is the signal intensity at equilibrium, and A is the saturation correction factor. Regional CBF was calculated from the formula CBF = λ·(T1obs·2α)-1·(MC – ML)·MC−1 where λ is the blood–brain partition coefficient of water (33), assuming a spatially constant value of 0.9 ml/g, and α is the spin-labeling efficiency measured in the carotids.
Measurement of PbO2
Cortical or thalamic PbO2 were measured at 24 h after 9 min asphyxial CA or sham surgery (n = 6–8 rats/group). The techniques for placement of the PbO2 electrode were described in a previous publication from our laboratory (3). Briefly, the head was stabilized in a stereotaxic instrument using ear bars. A small burr hole (2 mm) was drilled in the skull 2 mm lateral and 3.3 mm posterior to bregma. PbO2 was measured continuously using a Clark type tissue electrode (Ox-50, Unisense, Denmark). The electrode was inserted at a depth of 1 mm for cortical PbO2 measurement and 6 mm for thalamic PbO2 measurement as detailed below. We measured PbO2 in the cortex and thalamus, the two brain regions with the lowest or highest post-resuscitation CBF, respectively, in our previous report (2). Baseline parameters for MAP, PaO2, pH, PaCO2, and PbO2 were obtained after an equilibration period for PbO2 and before the CA.
Regional PbO2 Measurement at Different Oxygen Concentrations
We assessed cortical and thalamic PbO2 at FiO2 of 0.21, 0.5, and 1 after resuscitation from asphyxial CA. We chose the following sequence for FiO2 change to minimize electrode manipulations and FiO2 changes: the brain tissue oxygen electrode was inserted in the cortex while the rats received FiO2 = 0.5. Cortical PbO2 was measured at FiO2 = 0.5, FiO2 = 1.0, and FiO2 = 0.21, respectively. Next, the electrode was lowered into the thalamus, and the PbO2 was measured at FiO2 = 0.21, FiO2 = 1.0, and then FiO2 = 0.5, respectively. At each FiO2, we recorded the PbO2 and MAP after a 15 min stabilization period. Blood samples were obtained for arterial blood gas analysis. We aimed to have a representation of pH, PaO2, and PaCO2 values for each FiO2. Given the small size of the rats, small blood volume, combined with the possibility of hemodynamic instability after CA, we limited the repeated sampling of blood for the PbO2 studies. Each rat had a blood sample obtained at baseline and an additional sample obtained at either FiO2 = 0.21 or at FiO2 = 0.5.
Statistical Analysis
Data were analyzed with the statistical software Sigmastat 11.2 (Systat Software, San Jose, CA). CBF and PbO2 values after CA were compared to shams using the Mann-Whitney U-test with the Bonferroni correction for multiple comparisons (P < 0.01 for CBF and P < 0.016 for PbO2 analysis were considered significant).
Statement of Financial Support
This study was supported by: National Institutes of Health, Bethesda, MD R01HD075760 (M.D.M.), R01NS084604 (R.S.B.C.), Competitive Medical Research Fund, University of Pittsburgh, Pittsburgh, PA (M.D.M.).
Disclosure
None.
References
Moler FW, Meert K, Donaldson AE, et al.; Pediatric Emergency Care Applied Research Network. In-hospital versus out-of-hospital pediatric cardiac arrest: a multicenter cohort study. Crit Care Med 2009;37:2259–67.
Manole MD, Foley LM, Hitchens TK, et al. Magnetic resonance imaging assessment of regional cerebral blood flow after asphyxial cardiac arrest in immature rats. J Cereb Blood Flow Metab 2009;29:197–205.
Manole MD, Kochanek PM, Bayır H, et al. Brain tissue oxygen monitoring identifies cortical hypoxia and thalamic hyperoxia after experimental cardiac arrest in rats. Pediatr Res 2014;75:295–301.
Manole MD, Kochanek PM, Foley LM, et al. Polynitroxyl albumin and albumin therapy after pediatric asphyxial cardiac arrest: effects on cerebral blood flow and neurologic outcome. J Cereb Blood Flow Metab 2012;32:560–9.
Narotam PK, Morrison JF, Nathoo N. Brain tissue oxygen monitoring in traumatic brain injury and major trauma: outcome analysis of a brain tissue oxygen-directed therapy. J Neurosurg 2009;111:672–82.
Maloney-Wilensky E, Gracias V, Itkin A, et al. Brain tissue oxygen and outcome after severe traumatic brain injury: a systematic review. Crit Care Med 2009;37:2057–63.
Manole MD, Kochanek PM, Foley LM, et al. Polynitroxyl albumin and albumin therapy after pediatric asphyxial cardiac arrest: effects on cerebral blood flow and neurologic outcome. J Cereb Blood Flow Metab 2012;32:560–9.
Shaik JS, Poloyac SM, Kochanek PM, et al. 20-Hydroxyeicosatetraenoic acid inhibition by HET0016 offers neuroprotection, decreases edema, and increases cortical cerebral blood flow in a pediatric asphyxial cardiac arrest model in Rats. J Cereb Blood Flow Metab 2015;35:1757–63.
Choi SP, Park KN, Park HK, et al. Diffusion-weighted magnetic resonance imaging for predicting the clinical outcome of comatose survivors after cardiac arrest: a cohort study. Crit Care 2010;14:R17.
Radovsky A, Katz L, Ebmeyer U, Safar P. Ischemic neurons in rat brains after 6, 8, or 10 minutes of transient hypoxic ischemia. Toxicol Pathol 1997;25:500–5.
Böttiger BW, Schmitz B, Wiessner C, Vogel P, Hossmann KA. Neuronal stress response and neuronal cell damage after cardiocirculatory arrest in rats. J Cereb Blood Flow Metab 1998;18:1077–87.
Geocadin RG, Muthuswamy J, Sherman DL, Thakor NV, Hanley DF. Early electrophysiological and histologic changes after global cerebral ischemia in rats. Mov Disord 2000;15 Suppl 1:14–21.
Crick F. Function of the thalamic reticular complex: the searchlight hypothesis. Proc Natl Acad Sci USA 1984;81:4586–90.
McAlonan K, Cavanaugh J, Wurtz RH. Guarding the gateway to cortex with attention in visual thalamus. Nature 2008;456:391–4.
McCormick DA, Bal T. Sleep and arousal: thalamocortical mechanisms. Annu Rev Neurosci 1997;20:185–215.
Hartings JA, Simons DJ. Inhibition suppresses transmission of tonic vibrissa-evoked activity in the rat ventrobasal thalamus. J Neurosci 2000;20:RC100.
Venkatesan A, Frucht S. Movement disorders after resuscitation from cardiac arrest. Neurol Clin 2006;24:123–32.
Shoykhet M, Simons DJ, Alexander H, Hosler C, Kochanek PM, Clark RS. Thalamocortical dysfunction and thalamic injury after asphyxial cardiac arrest in developing rats. J Neurosci 2012;32:4972–81.
Aravamuthan BR, Shoykhet M. Long-term increase in coherence between the basal ganglia and motor cortex after asphyxial cardiac arrest and resuscitation in developing rats. Pediatr Res 2015;78:371–9.
Goodman JC, Valadka AB, Gopinath SP, Uzura M, Robertson CS. Extracellular lactate and glucose alterations in the brain after head injury measured by microdialysis. Crit Care Med 1999;27:1965–73.
Nordmark J, Rubertsson S, Mörtberg E, Nilsson P, Enblad P. Intracerebral monitoring in comatose patients treated with hypothermia after a cardiac arrest. Acta Anaesthesiol Scand 2009;53:289–98.
Hayashida K, Nishiyama K, Suzuki M, et al. Estimated cerebral oxyhemoglobin as a useful indicator of neuroprotection in patients with post-cardiac arrest syndrome: a prospective, multicenter observational study. Crit Care 2014;18:500.
Genbrugge C, Meex I, Boer W, et al. Increase in cerebral oxygenation during advanced life support in out-of-hospital patients is associated with return of spontaneous circulation. Crit Care 2015;19:112.
Bouglé A, Daviaud F, Bougouin W, et al. Determinants and significance of cerebral oximetry after cardiac arrest: A prospective cohort study. Resuscitation 2016;99:1–6.
Duggan M, McNamara PJ, Engelberts D, et al. Oxygen attenuates atelectasis-induced injury in the in vivo rat lung. Anesthesiology 2005;103:522–31.
Atkins JL, Johnson KB, Pearce FJ. Cardiovascular responses to oxygen inhalation after hemorrhage in anesthetized rats: hyperoxic vasoconstriction. Am J Physiol Heart Circ Physiol 2007;292:H776–85.
Jackson WF. Arteriolar oxygen reactivity: where is the sensor? Am J Physiol 1987;253(5 Pt 2):H1120–6.
Fink EL, Alexander H, Marco CD, et al. Experimental model of pediatric asphyxial cardiopulmonary arrest in rats. Pediatr Crit Care Med 2004;5:139–44.
Hendrich KS, Kochanek PM, Melick JA, et al. Cerebral perfusion during anesthesia with fentanyl, isoflurane, or pentobarbital in normal rats studied by arterial spin-labeled MRI. Magn Reson Med 2001;46:202–6.
Detre JA, Leigh JS, Williams DS, Koretsky AP. Perfusion imaging. Magn Reson Med 1992;23:37–45.
Hendrich KS, Kochanek PM, Williams DS, Schiding JK, Marion DW, Ho C. Early perfusion after controlled cortical impact in rats: quantification by arterial spin-labeled MRI and the influence of spin-lattice relaxation time heterogeneity. Magn Reson Med 1999;42:673–81.
Zhang W, Williams DS, Koretsky AP. Measurement of rat brain perfusion by NMR using spin labeling of arterial water: in vivo determination of the degree of spin labeling. Magn Reson Med 1993;29:416–21.
Herscovitch P, Raichle ME. What is the correct value for the brain–blood partition coefficient for water? J Cereb Blood Flow Metab 1985;5:65–9.
Author information
Authors and Affiliations
Corresponding author
PowerPoint slides
Rights and permissions
About this article

Cite this article
Foley, L., Clark, R., Vazquez, A. et al. Enduring disturbances in regional cerebral blood flow and brain oxygenation at 24 h after asphyxial cardiac arrest in developing rats. Pediatr Res 81, 94–98 (2017). https://doi.org/10.1038/pr.2016.175
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/pr.2016.175
This article is cited by
-
Early Thalamocortical Reperfusion Leads to Neurologic Recovery in a Rodent Cardiac Arrest Model
Neurocritical Care (2022)
-
Neonatal cerebrovascular autoregulation
Pediatric Research (2018)
