
Abstract
Chronic fetal anemia results in significant cardiac remodeling. The capacity to reverse these effects is unknown. We examined the effects of transfusion on cardiomyocyte adaptations after chronic anemia in fetal sheep subjected to daily hemorrhage beginning at 109-d GA (term ∼145 d). After 10 d of anemia, one group was killed for comparison with age-matched controls. A separate group of anemic fetuses was transfused with red blood cells at 119-d GA for comparison with controls at 129-d GA. Anemia significantly increased the heart-to-body weight ratio, an effect partially ameliorated after transfusion. Cardiomyocyte dimensions were similar among all groups, suggesting an absence of hypertrophy. The percentages of mono- and binucleated cardiomyocytes were similar between groups at 119-d GA, although the percentage of binucleated cells was significantly less in transfused fetuses compared with controls at 129-d GA. Protein levels of mitogen-activated protein kinases and protein kinase B were similar between controls and their respective intervention groups, except for a significant increase in phosphorylated c-Jun N-terminal kinase 1/2 in transfused fetuses. Thus, cardiomyocyte proliferation but not hypertrophy contributes to cardiac enlargement during fetal anemia. Transfusion results in slowing but not cessation of cardiac growth after anemia.
Similar content being viewed by others
Main
Chronic fetal anemia imparts a significant challenge on the cardiovascular system to maintain systemic oxygen delivery. Adaptations in the fetus include marked increases in cardiac output and cardiac mass (1,2). The hemodynamic stress of anemia potentially leads to congestive heart failure, hydrops fetalis, and fetal demise (3). In the clinical setting, transfusion of the anemic fetus with red blood cells is the standard therapy (4), but treatment may fail to mitigate cardiac enlargement as determined at birth (2). Fetal cardiac growth is remarkably plastic, adapting to cardiovascular stress differently than the mature, postnatal heart. In the adult heart, cardiac myocytes are multinucleated and classically believed to be nonproliferative with cardiac growth occurring primarily by cardiomyocyte hypertrophy. In contrast, cardiomyocytes actively proliferate during normal growth in the fetal heart (5). Cellular enlargement and the transition to terminal differentiation are also important to heart growth during the perinatal period, as are coordinated expansion of vascular and connective tissues. In the near-term fetus, anemia causes cardiac enlargement by accelerating normal growth processes, including cardiomyocyte hyperplasia and hypertrophy (6,7). The mitogen-activated protein kinase (MAPK) signaling pathways are likely important regulators of these adaptations, although there may be important differences in activation based on developmental stage (8).
Given the adaptability of the fetal heart in response to stress, it may be predicted that the immature heart would respond readily to resolution of that stress. This study was designed to test the hypothesis that, in the enlarged hearts of anemic fetuses, red blood cell transfusion and restoration of Hb would result in transient cessation of cardiomyocyte growth and proliferation, with normalization of heart weight and cardiomyocyte dimensions. Given their purported role in regulating cardiac growth, we also examined changes in the expression of terminal MAPK proteins and protein kinase B (AKT). We have shown previously that with induction of anemia as outlined in this study, the hearts of anemic fetuses grow to be ∼40% heavier than age-matched controls (7). According to our growth curves, the fetal sheep heart is ∼40% heavier at 129-d GA than at 119-d GA (5). Therefore, this 10-d recovery period was selected to test the hypothesis that the heart-to-body weight ratio would normalize by cardiac growth arrest after transfusion.
METHODS
Animal experiments were approved by the University of Iowa Institutional Animal Care and Use Committee and were conducted within the regulations of the Animal Welfare Act and the National Research Council's Guide for the Care and use of Laboratory Animals. Gestation-timed ewes of mixed western breed were obtained from a local supplier and acclimatized to the laboratory. At 103-d GA (term ∼145 d), catheterization of the fetus was performed. After an intramuscular injection of 7.5 mg thiopental sodium (12 mg/kg; Abbott Laboratories, Chicago, IL) was used to induce anesthesia, which was maintained with isoflurane (2%) in a 30/70 mixture of oxygen/nitrous oxide. The uterus was exposed through a midline incision, and the fetal head exteriorized. Indwelling catheters were placed in a fetal carotid artery and jugular vein (0.86 or 1.19 mm internal diameter; Scientific Commodities Incorporated, Lake Havasu City, AZ). An amniotic fluid catheter (1.19 mm internal diameter) was anchored to the fetal skin. Fetal, uterine, and maternal flank incisions were closed in separate layers, and catheters exteriorized through a s.c. tunnel and stored in a cloth pouch on the ewe's flank. At the end of the surgery, and for 3 d postoperatively, ampicillin was infused into the amniotic cavity through the amniotic fluid catheter (2 g) and given i.m. to the ewe (2 g). Butorphanol (0.1 mg/kg i.v.; Torbugesic; Fort Dodge Animal Health, Fort Dodge, IA) was given for 24 h postoperatively for analgesia. Ewes and their fetuses were allowed to recover for 6 d before beginning experiments.
Physiological studies.
Fetuses were alternately allocated to one of four groups: five control fetuses were not bled and were killed after 10 d of study (control); six fetuses were bled to reduce their arterial oxygen content and were killed after 10 d of study (anemic); seven control fetuses were not bled and were killed after 20 d of study (control-recovery); seven fetuses were bled to reduce their arterial oxygen content for 10 d; on the 10th day, they were transfused with autologous packed red blood cells to restore their hematocrit toward control values. These animals were killed after 20 d of study (anemic-recovery). All experiments began at 109-d GA with the control and anemic experiments concluded at 119-d GA and the control-recovery and anemic-recovery experiments concluded at 129-d GA.
The experimental protocol was initiated at 109-d GA (day 0) with measurement of baseline hemodynamics, arterial blood pH, PO2, PCO2 (Gem Premier 3000, Instrumentation Laboratory, Bedford, MA), hematocrit, Hb, and oxygen content (IL 682 co-oximeter system; Instrumentation Laboratory). Arterial pressure was measured with Transpac pressure transducers (Abbott, Abbott Park, IL) connected to a calibrated PowerLab computerized recording system (ADInstruments, CO Springs, CO). Arterial pressure was referred to amniotic fluid pressure and reported as the arithmetic mean from computer tracings. Arterial pressure tracings were used to calculate fetal heart rate. Hemodynamics were recorded at baseline and on day 10 of the study. In the anemia-recovery and control-recovery groups, hemodynamics were also recorded for 2 d after transfusion (days 11 and 12) and on the final experimental day (day 20).
Fetuses assigned to an anemia group had blood removed according to a formula determined empirically during previous studies in the laboratory to rapidly induce anemia with minimal acute fetal demise, modified for the age (and lower total blood volume) of the fetuses in this study (7). In the anemic groups, an average of 69 ± 19 mL (mean ± SD) of blood was removed daily for the first 3 d (until arterial oxygen content was less than 4 mg/dL); thereafter, 39 ± 19 mL of blood was removed daily. Blood was collected in a commercially available blood collection system, and the red blood cells separated into a RBC storage solution according to the manufacture's protocol (Optisol Triple Blood Bag system; Terumo Transfusion Solutions, Somerset, NJ). The volume of blood was replaced daily with an equivalent volume of warmed 0.9% NaCl solution. Control fetuses were not hemorrhaged. On the tenth day, fetuses in the anemic-recovery group were transfused with warmed, autologous packed red blood cells after passage through an i.v. transfusion filter. If fetal arterial pressure started to rise, transfusion was terminated and resumed the following day (on average, 66% of the transfusion occurred on day 10). The total blood volume transfused to each anemic-recovery fetus was 56 ± 20 mL.
Postmortem.
At the conclusion of the experimental period, ewes were killed with sodium pentobarbital (65 mg/kg i.v.). Deeply anesthetized fetuses were given heparin (5000 U i.v.) and saturated potassium chloride (10 mL i.v.) to arrest their hearts in diastole. Fetuses and their excised hearts were weighed.
Cardiac dissociation and cardiomyocyte analysis.
A full-thickness transverse section of the left ventricular (LV) free-wall was excised and frozen in liquid nitrogen. The wound edges were sealed with cyanoacrylate. Fetal hearts were then enzymatically dissociated on a Langendorff apparatus, and the cardiomyocytes were fixed for morphometric analysis as previously described (5). Myocytes were photographed at 40× on a light microscope (Zeiss Axiophot; Bartels and Stout, Bellevue, WA), and photomicrographs analyzed by calibrated software (Image Pro Plus; MediaCybernetics, Bethesda, MD). At least 50 cells of each type (mononucleated or binucleated) were measured per ventricle per fetus. Separately, at least 300 myocytes from each ventricle of each fetus were counted to determine the number of nuclei per cardiomyocyte.
Cell cycle activity.
The anti-Ki-67 antibody MIB-1 (DAKO, Carpinteria, CA) was used to immunohistologically detect cell cycle activity in dissociated cardiomyocytes as previously described (9). Detection of antibody binding was carried out using an avidin–biotin system (Vectastain ABC kit; Vector Laboratories, Burlingame, CA) and staining with diaminobenzidine. No fewer than 500 myocytes were counted per ventricle per fetus for cell cycle activity analysis. Ki-67 positive myocytes are expressed as a percent of total mononucleated cardiomyocytes.
Quantitative immunoblot analysis.
Immunoblots were prepared as described previously (8). Briefly, myocardial samples were homogenized in the presence of protease inhibitors, centrifuged, and total protein quantitated spectrophotometrically. Twenty microgram of protein were separated by SDS-PAGE and transferred to a nitrocellulose membrane. Membranes were blocked with Odyssey Blocking Buffer (Li-Cor Biosciences, Lincoln, NE) for 1 h at room temperature and then incubated in primary antibody overnight at 5°C. Bound primary antibody was detected by incubation with infrared-labeled secondary antibodies (IRDye 800 or IRDye 700 700DX; Li-Cor Biosciences) and quantitated with a Li-Cor Odyssey Imaging System. Primary antibodies obtained from Santa Cruz Biotechnology (Santa Cruz, CA) include total extracellular signal regulated kinase (ERK) 1/2 (1:1000 dilution, sc-93); phosphorylated ERK1/2 (1:1000, sc-7383); total c-Jun N-terminal kinase (JNK) 1/2 (1:1000, sc-1648); phosphorylated JNK1/2 (1:1000, sc-6254), and total p38 (1:1000, sc-9212); and from Cell Signaling Technology (Beverly, MA): phosphorylated p38 (1:250, No. 9122); and AKT (1:1000, No. 9272).
Statistical analysis.
Daily hemodynamic and blood gas data were compared within groups by one-way ANOVA. If warranted by the F statistic, Dunnett's multiple comparison was performed as a posttest to determine what days were different from baseline. Data were also compared among groups on day 10 by one-way ANOVA, and if warranted, Bonferroni's multiple comparison test was performed to identify differences. On the final experimental day, hemodynamic and blood gas data were compared between groups by unpaired t test. Body and organ weights, cardiomyocyte dimensions, terminal differentiation, and cell cycle activity were compared by one-way ANOVA and, if warranted, by Bonferroni's multiple comparison test. Protein data were compared by unpaired t test. All data are presented as mean ± SEM. A value of p < 0.05 was accepted as statistically significant.
RESULTS
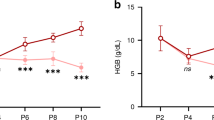
Bleeding significantly reduced fetal hematocrit below baseline values by the second (anemic-recovery) and third (anemic) days (Fig. 1A). In both anemic groups, hematocrit was significantly lower than the matched control group (control and control-recovery) on days 3–10. After transfusion of the anemia-recovery group, hematocrit remained lower than baseline on day 11 (first day posttransfusion) but subsequently returned toward baseline (day 0). Hematocrit also returned to values similar to those in the control-recovery group.

Arterial blood gas and hemodynamic values. (A) Hematocrit; (B) total Hb; (C) O2 content; (D) partial pressure of O2; (E) partial pressure of CO2; (F) pH; (G) mean arterial blood pressure; and (H) heart rate. Control: •; control-recovery: ▪; anemic: ○; anemic-recovery: □. Significantly different (p < 0.05) from baseline (experimental day 0) for *anemic group; †anemic-recovery group; ‡control-recovery group; §control group. **Anemic group significantly different from control group (p < 0.05); ¶anemic-recovery group significantly different from control-recovery group (p < 0.05).
Total Hb and arterial oxygen content were also reduced on the second (anemic-recovery) and third (anemic) days compared with baseline (Fig. 1B and C). Hb and arterial oxygen content in both anemic groups remained significantly less than the matched control group on days 3–10. After transfusion of the anemic-recovery group, total Hb remained slightly but significantly less from baseline on days 11, 12, and 14, whereas arterial oxygen content remained different from baseline until day 18. Arterial PO2 and PCO2 were relatively unchanged by bleeding in either anemic group (Fig. 1D and E). A slight but statistically significant reduction in pH was detected in the anemic-recovery group on multiple study days compared with baseline, although no differences between groups were detected (Fig. 1F).
Mean arterial blood pressure was similar among all groups at baseline and day 10 (Fig. 1G). Arterial pressure was also unaffected by transfusion (days 11 and 12). On the final experimental day, both the anemic-recovery and control-recovery groups had elevated arterial pressures when compared with baseline, as would be expected with increasing gestation (10). Heart rate, which also tends to decline with advancing GA, was lower than baseline in the control group on day 10 and in the anemic-recovery group on day 20 (Fig. 1H).
Fetal body weights were not different between anemic groups and their age-matched control groups at either 119-d GA (anemic) or 129-d GA (anemic-recovery; Fig. 2A), although as expected, body weights were increased at 129-d GA compared with 119-d GA. Heart weight was 45% greater in the anemic group compared with control group (22.58 ± 1.31 versus 15.58 ± 0.91 g, p < 0.05; Fig. 2B). At 129-d GA, mean heart weight from anemic-recovery fetuses (28.31 ± 1.32 g) was still significantly greater than from recovery-control (24.84 ± 1.31 g), although the percentage increase (14%) was less than that present between anemic and control groups at 119-d GA. Heart weights were also significantly increased at 129-d GA compared with 119-d GA when compared within treatment. When expressed relative to body weight, heart weight in both anemic and anemic-recovery groups was significantly greater than their age-matched controls (Fig. 2C).

Fetal body and heart weights. (A) Body weight; (B) heart weight; (C) heart-to-body weight ratio. Significantly different (p < 0.05) from *age-matched control group; †treatment-matched group.
The percentage of binucleated cardiomyocytes (an index of terminal differentiation) was not different between the anemic and control groups in either ventricle (Fig. 3A and B). Within the control groups, there was an increase in cardiomyocyte binucleation from 119-d GA to 129-d GA, consistent with the normal maturational process. However, this increase was not seen in the anemic groups, with binucleation similar in anemic (119-d GA) and anemic-recovery groups (129-d GA) in both ventricles. Furthermore, the percentage of binucleated cardiomyocytes was significantly greater in control-recovery than in anemic-recovery hearts in both ventricles. Cell cycle activity (as determined by Ki-67 immunostaining of mononucleated cells) was similar among all groups in both the LV and right ventricle (RV) (Fig. 3C and D). Evidence of cardiomyocyte hypertrophy was assessed by examination of fixed, isolated cardiomyocyte length and width. No significant differences in cardiomyocyte dimensions were detected among groups (Table 1).

Fetal cardiomyocyte maturation and cell cycle activity. Cell cycle activity is represented by the %Ki-67 positive mononucleated cardiomyocytes. (A) LV binucleation; (B) RV binucleation; (C) LV cell cycle activity; (D) RV cell cycle activity. †p < 0.05 compared with treatment-matched group.
LV protein levels of MAPK pathway components were determined in a subset of the 129-d GA groups (Fig. 4). Total protein levels of, ERK, JNK, p38, and AKT were similar between the control-recovery and anemic-recovery groups. Similarly, phospho-ERK, phospho-p38, and phospho-AKT levels were not different between these groups. However, myocardial levels of phospho-JNK were significantly greater in the anemic-recovery compared with the control-recovery group.

Myocardial MAPK and AKT expression. Steady-state protein levels of total (A, C, E, and G) and activated (phosphorylated; B, D, F, and H) ERK1/2, JNK, p38, and AKT. *p < 0.05 compared with control levels in anemic-recovery hearts. Phospho-p38 tended to be higher in the anemic-recovery hearts (p = 0.078); n = 6–7 for each group.
DISCUSSION
Adaptive cardiac growth enables the fetus to meet the challenges of chronic anemia. Peripheral tissues respond to progressive anemia by demanding more blood flow, and cardiac enlargement in the chronically anemic fetus supports a sustained increase in cardiac output (11). Although changes in cardiomyocyte morphology, metabolism, and cardiac vascularity (6,12–14) accompany cardiac growth in response to chronic fetal anemia, it is unclear whether any of these changes are reversible on resolution of anemia. We approached this problem by examining the cardiomyocyte response after transfusion of autologous red blood cells to anemic fetuses. Our results suggest that myocardial growth continues after transfusion of the anemic fetus, albeit more slowly than normal for gestation age. Slowed growth may be mediated by a delay of the cardiomyocyte maturational process, as evidenced by the decreased percentage of binucleated cardiomyocytes and/or activation of JNK1/2, which has been shown to antagonize cardiac growth in other animal models (15). However, cardiac growth appears to continue after transfusion, indicating that mitogenic signals dominate the signaling milieu of the fetal myocardium. Thus, although transfusion obviates the need for the adaptive cardiac functional response, excess cardiac capacity does not completely inhibit continued heart growth.
Cardiomyocyte responses.
In late gestation sheep fetuses (129–138-d GA), cardiac enlargement in response to chronic anemia is accomplished by balanced growth of all components of the myocardium, including cardiomyocyte proliferation and hypertrophy (7). In these studies, the percentage of binucleated cardiomyocytes increased in the RV of anemic fetuses, presenting a picture of a more mature heart. In this study, in which fetuses were made anemic at a much younger age (beginning 109-d GA), the mechanisms of increased cardiac mass are unclear, although cardiomyocyte proliferation was likely responsible for the majority of the substantial cardiac enlargement as we did not detect evidence of cardiomyocyte hypertrophy (by cell dimensions) or maturation (by percentage binucleation). Reasons for the differences in response likely relate to maturational differences in the heart at the initiation of the study. For example, at 109-d GA, the fetal heart is composed almost entirely of mononucleated cardiomyocytes, capable of proliferation and/or terminal differentiation into binucleated cells (5). However, by 129-d GA, the composition of the heart includes only ∼60% mononucleated cells. The nature of the mechanisms regulating this maturational process, including terminal differentiation and its independence or interdependence on pathways stimulating cell cycle activity and proliferation, remains to be explored.
Although we failed to see evidence of increased Ki-67 immunostaining, a marker of cell cycle activity, we speculate that, after 10 d of chronic anemia, the cardiomyocyte proliferative response had ceased as further cardiac adaptation to anemia was not needed (7). This lack of sustained cardiomyocyte proliferation is in sharp contrast to a recent study by our group, which found that, after 5 d of chronic anemia and before the onset of a significant increase in myocardial mass, there were twice as many Ki-67 positive stained cardiomyocytes in anemic compared with control fetal hearts (16). Thus, we expect that, at the end of the 10 d anemic period, the hearts of these fetuses contained many more cardiomyocytes than normal for their age, although the percentage of binucleated and mononucleated cells within each ventricle are similar to control.
Because we did not perform a longitudinal study and follow-up cardiac mass over time within each animal, it is difficult to fully assess the effect of transfusion on subsequent heart growth. There did not appear to be complete cessation of cardiac growth after transfusion, as this would have resulted in normalization of heart weights between anemic-recovery and control-recovery groups. However, transfusion of anemic fetuses led to an apparent substantial slowing of heart growth. This reduced rate of growth was evident by the hearts from anemic fetus at 119-d GA being on average 45% greater in mass than their respective controls, whereas hearts from transfused fetuses were only 14% greater in mass than controls, with a rate of increase of 0.93 g/d in control animals and 0.57 g/d in anemic fetuses. In addition, heart weight/body weight in anemic fetuses tended to decrease from 8.7 ± 0.4 g/kg to 8.0 ± 0.5 g/kg after transfusion (p = 0.34). Taken together, these data suggest that transfusion of red cells to anemic fetuses whose hearts have undergone adaption to increase cardiac output and maintain oxygen delivery to peripheral tissues retards but does not eliminate age-dependent myocardial growth. This is consistent with the finding that intrauterine transfusion imperfectly resolved the cardiac enlargement due to fetal anemia in human pregnancy (2).
Intracellular signaling pathways.
Mechanisms mediating the adaptive response of the fetal heart to chronic anemia have been explored. Martin et al. (6) found that the increase in cardiac mass and vascularization resulting from chronic anemia was associated with induction of hypoxia inducible factor (HIF)-1 and VEGF). By using this same model, we subsequently described increases in myocardial expression of VEGF receptors and a number of HIF-1 regulated glycolytic enzymes (13). The MAPK signaling pathways have also been explored in this fetal sheep model given their important roles in cardiac growth and hypertrophy (8). In genetically altered mice, concentric, compensated cardiac hypertrophy results from the overexpression of active MAPK kinase (MEK)1, which activates ERK, whereas dilated cardiomyopathy and premature death are reported in mice with overactivation of p38 or JNK (17,18). In our sheep studies, we found the MAPK response to chronic anemia was dependent on the gestation age of the animal, despite similar increase in cardiac mass (8). Specifically, young fetuses (∼100 d gestation) had no significant changes in myocardial expression of phosphorylated ERK, JNK, or p38 in response to anemia, whereas older fetuses (∼130 d gestation) had a marked decrease in ERK but not JNK or p38 phosphorylation. In this study, we again found no significant changes in myocardial MAPK or AKT activities in response to anemia. Given the nature of the chronic growth stimulus (anemia), we recognize that changes in activity of these regulatory pathways may have occurred early with the anemic stimulus but returned to baseline by the end of the study when tissue sampling occurred. Thus, it remains possible that the proliferative cardiomyocyte growth in young chronically anemic fetal sheep (which necessarily occurred given the increase in mass in the absence of cellular hypertrophy) is the result of increased activity of MAPK and/or AKT pathways.
In contrast to the control or anemic groups, we found that JNK phosphorylation was increased in the anemic-recovery group in this study, consistent with its role for inhibiting myocardial growth (15). Phosphorylation of p38 also tended to be increased in the anemic-recovery group, although the difference was not statistically significant. These data suggest that the fetal heart responds to resolution of anemic stress by activating growth inhibitory pathways. Cardiac growth nevertheless continued during the recovery period, indicating that promitogenic signals continued to dominate the signaling milieu of the fetal myocardium.
Limitations.
The red cell volume transfused back to fetuses was insufficient to return hematocrit, total Hb, and arterial oxygen content entirely to prehemorrhage levels. This may be due to destruction of some red blood cells during handling, as well as the increase in total circulating blood volume that accompanies fetal growth. It is undetermined if this small difference in oxygen content was the stimulus for continued heart growth after transfusion or if other mitogenic signals were responsible.
Changes in extracellular matrix and vascularity were not examined and could contribute to the increased cardiac mass in anemic animals. However, Jonker et al. (7) previously demonstrated in chronically anemic fetuses that the ventricular tissue fraction of collagen, other connective tissue, and vascular space did not differ between anemic and control fetuses. On the other hand, fetal cardiac vascular adaptations to chronic anemia, including increased capillary volume density and diameter and decreased intercapillary distance, have been described (6,13). More detailed examination of these cardiac components and their adaption to anemia and transfusion is warranted.
Although our discussion of cardiomyocyte maturation has focused on nucleation (i.e. mononucleated cells being immature), it must be noted that mononucleated cells may become polyploid, and thus terminally differentiated, particularly in response to hemodynamic stress (19). Although we did not assess cellular ploidy, such changes, if present, may suggest that chronic anemia alters the maturation of the fetal heart and account for the lack of observed effect on cardiomyocyte proliferation.
Perspective.
Anemia is a powerful stimulus for accelerated growth of the immature fetal heart. Systemic oxygen demand drives the need for increased cardiac output, which is achieved by an increase in stroke volume resulting from expanding the ventricular chamber volume. This cardiac remodeling includes increased ventricular wall thickness to reduce wall stress, according to the Law of Laplace. Although restoration of hematocrit by transfusion reduces the demand for cardiac output, correction of anemia by red blood cell transfusion does not arrest growth of the enlarged fetal heart. Activation of antigrowth MAPK pathways (JNK) suggests that the fetal heart responded to changing hemodynamic conditions, but even in the context of this recovery period, the signaling milieu of the fetal myocardium remained progrowth. Thus, factors other than demand for cardiac output likely drive basal heart growth in the growing fetus.
The ultrastructure of the heart is likely a primary factor in maintaining cardiac health through the lifespan. Cumulative loss of cardiomyocytes contributes to progression of cardiac disease, thus an increased cardiomyocyte endowment at birth may be beneficial. Fetal anemia compromises pregnancy and may lead to fetal demise, but the long-term cardiac risks for surviving infants are not clear. Adult sheep made anemic in utero and transfused to normal hematocrit before birth has an enhanced contractile response during hypoxia (20) but increased susceptibility to ischemia-reperfusion injury (21). The extent to which these differences are the result of altered coronary anatomy and physiology or augmented cardiomyocyte endowment is unknown. Nevertheless, disruption of the anatomical and physiological relationships between vasculature and cardiomyocyte may have serious consequences. Further research is indicated to determine therapies additional to transfusion to mitigate the long-term cardiac outcomes of fetal anemia.
Abbreviations
- AKT:
-
protein kinase B
- ERK:
-
extracellular signal regulated kinase
- JNK:
-
c-Jun N-terminal kinase
- LV:
-
left ventricle
- MAPK:
-
mitogen-activated protein kinase
- RV:
-
right ventricle
REFERENCES
Sivasankaran S, Sharland GK, Simpson JM 2005 Dilated cardiomyopathy presenting during fetal life. Cardiol Young 15: 409–416
Carter BS, DiGiacomo JE, Balderston SM, Wiggins JW, Merenstein GB 1990 Disproportionate septal hypertrophy associated with erythroblastosis fetalis. Am J Dis Child 144: 1225–1228
Huhta JC 2004 Guidelines for the evaluation of heart failure in the fetus with or without hydrops. Pediatr Cardiol 25: 274–286
Moise KJ Jr 1993 Intrauterine transfusion with red cells and platelets. West J Med 159: 318–324
Jonker SS, Zhang L, Louey S, Giraud GD, Thornburg KL, Faber JJ 2007 Myocyte enlargement, differentiation, and proliferation kinetics in the fetal sheep heart. J Appl Physiol 102: 1130–1142
Martin C, Yu AY, Jiang BH, Davis L, Kimberly D, Hohimer AR, Semenza GL 1998 Cardiac hypertrophy in chronically anemic fetal sheep: increased vascularization is associated with increased myocardial expression of vascular endothelial growth factor and hypoxia-inducible factor 1. Am J Obstet Gynecol 178: 527–534
Jonker SS, Giraud MK, Giraud GD, Chattergoon NN, Louey S, Davis LE, Faber JJ, Thornburg KL 2010 Cardiomyocyte enlargement, proliferation, and maturation during chronic fetal anemia in sheep. Exp Physiol 95: 131–139
Olson AK, Protheroe KN, Scholz TD, Segar JL 2006 The mitogen-activated protein kinases and Akt are developmentally regulated in the chronically anemic fetal sheep heart. J Soc Gynecol Investig 13: 157–165
Jonker SS, Faber JJ, Anderson DF, Thornburg KL, Louey S, Giraud GD 2007 Sequential growth of fetal sheep cardiac myocytes in response to simultaneous arterial and venous hypertension. Am J Physiol Regul Integr Comp Physiol 292: R913–R919
Unno N, Wong CH, Jenkins SL, Wentworth RA, Ding XY, Li C, Robertson SS, Smotherman WP, Nathanielsz PW 1999 Blood pressure and heart rate in the ovine fetus: ontogenic changes and effects of fetal adrenalectomy. Am J Physiol 276: H248–H256
Davis LE, Hohimer AR 1991 Hemodynamics and organ blood flow in fetal sheep subjected to chronic anemia. Am J Physiol 261: R1542–R1548
Davis LE, Hohimer AR, Morton MJ 1999 Myocardial blood flow and coronary reserve in chronically anemic fetal lambs. Am J Physiol 277: R306–R313
Mascio CE, Olison AK, Ralphe JC, Tomanek RJ, Scholz TD, Segar JL 2005 Myocardial vascular and metabolic adaptations in chronically anemic fetal sheep. Am J Physiol Regul Integr Comp Physiol 289: R1736–R1745
Ralphe JC, Nau PN, Mascio CE, Segar JL, Scholz TD 2005 Regulation of myocardial glucose transporters GLUT1 and GLUT4 in chronically anemic fetal lambs. Pediatr Res 58: 713–718
Liang Q, Bueno OF, Wilkins BJ, Kuan CY, Xia Y, Molkentin JD 2003 c-Jun N-terminal kinases (JNK) antagonize cardiac growth through cross-talk with calcineurin-NFAT signaling. EMBO J 22: 5079–5089
Jonker SS, Scholz TD, Segar JL 2011 The effect of adrenalectomy on the cardiac response to subacute fetal anemia. Can J Physiol Pharmacol 89: 79–88
Bueno OF, De Windt LJ, Tymitz KM, Witt SA, Kimball TR, Klevitsky R, Hewett TE, Jones SP, Lefer DJ, Peng CF, Kitsis RN, Molkentin JD 2000 The MEK1-ERK1/2 signaling pathway promotes compensated cardiac hypertrophy in transgenic mice. EMBO J 19: 6341–6350
Liao P, Georgakopoulos D, Kovacs A, Zheng M, Lerner D, Pu H, Saffitz J, Chien K, Xiao RP, Kass DA, Wang Y 2001 The in vivo role of p38 MAP kinases in cardiac remodeling and restrictive cardiomyopathy. Proc Natl Acad Sci USA 98: 12283–12288
Bensley JG, Stacy VK, De Matteo R, Harding R, Black MJ 2010 Cardiac remodelling as a result of pre-term birth: implications for future cardiovascular disease. Eur Heart J 31: 2058–2066
Broberg CS, Giraud GD, Schultz JM, Thornburg KL, Hohimer AR, Davis LE 2003 Fetal anemia leads to augmented contractile response to hypoxic stress in adulthood. Am J Physiol Regul Integr Comp Physiol 285: R649–R655
Yang Q, Hohimer AR, Giraud GD, Van Winkle DM, Underwood MJ, He GW, Davis LE 2008 Effect of fetal anaemia on myocardial ischaemia-reperfusion injury and coronary vasoreactivity in adult sheep. Acta Physiol (Oxf) 194: 325–334
Author information
Authors and Affiliations
Corresponding author
Additional information
Supported by Grant R01HL080657 [J.L.S.] and Grants F32HL088787, L40HL097627-01, and Oregon BIRCWH K12HD043488 [S.S.J.].S.S.J. is currently at Oregon Health & Science University.
Rights and permissions
About this article
Cite this article
Jonker, S., Scholz, T. & Segar, J. Transfusion Effects on Cardiomyocyte Growth and Proliferation in Fetal Sheep After Chronic Anemia. Pediatr Res 69, 485–490 (2011). https://doi.org/10.1203/PDR.0b013e3182181e01
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/PDR.0b013e3182181e01
