
Abstract
Premature infants are at increased risk of developing airway hyper-reactivity (AHR) after oxidative stress and inflammation. Mast cells contribute to AHR partly by mediator release, so we sought to determine whether blocking mast cell degranulation or recruitment prevents hyperoxia-induced AHR, mast cell accumulation, and airway smooth muscle (ASM) changes. Rats were exposed at birth to air or 60% O2 for 14 d, inducing significantly increased AHR in the latter group, induced by nebulized methacholine challenge and measured by forced oscillometry. Daily treatment (postnatal d 1–14) with intraperitoneal cromolyn prevented hyperoxia-induced AHR, as did treatment with imatinib on postnatal d 5–14, compared with vehicle treated controls. Cromolyn prevented mast cell degranulation in the trachea but not hilar airways and blocked mast cell accumulation in the hilar airways. Imatinib treatment completely blocked mast cell accumulation in tracheal/hilar airway tissues. Hyperoxia-induced AHR in neonatal rats is mediated, at least in part, via the mast cell.
Similar content being viewed by others


Main
Bronchopulmonary dysplasia (BPD) contributes to increased mortality and long-term morbidity in premature infants and is associated with increased airway hyper-reactivity (AHR) (1). Even brief postnatal exposures to supplemental oxygen are associated with impaired airway function and AHR measured in early adulthood (2). The mechanisms that contribute to BPD-associated AHR have yet to be firmly established.
The available evidence suggests that BPD-associated wheezing is distinct from allergic asthma in several respects. The lungs in infants with BPD are characterized by dysplastic airway epithelium, interstitial edema, and impaired alveolarization (3). Children with wheezing after BPD do not exhibit increased exhaled nitric oxide, which is found in higher levels in asthmatics secondary to persistent airway inflammation (4). Survivors of BPD are less likely to show signs of atopy and have fewer eosinophils in their sputum compared with asthmatics (2). There is limited information about the structural differences between the airways of patients with allergic asthma and those of patients with AHR after BPD, but it is generally agreed that they are less responsive to bronchodilators than allergic asthmatics (4).
Despite these phenotypical differences, inflammatory mechanisms are common to both conditions and are targets of therapeutic interventions. Mast cell functions may play a role in both conditions. They contribute to allergy-induced AHR through release of histamine, cytokines, and proteases that result in airway smooth muscle (ASM) contraction, increased vascular permeability, and leukocyte migration (5). Increased mast cell numbers have been observed in BPD-affected lungs in baboons (6) and humans (7). Hyperoxia-exposure increases tracheal mast cell accumulation in newborn rats (8).
We hypothesized that mast cells may contribute significantly to oxidative stress and induction of AHR in newborns, and to test this hypothesis, we used an established newborn rat model of BPD, which produces patchy alveolar hypoplasia (9). We found that hyperoxia-induced mast cell accumulation and methacholine challenge-induced AHR. Treatment with systemic cromolyn prevented pulmonary mast cell degranulation and methacholine-induced AHR. Systemic treatment with imatinib mesylate, a known inhibitor of mast cell homing (10), decreased mast cell abundance and hyperoxia-induced AHR. Some of the data have been reported in abstract form (E. Schultz et al., Cromolyn blocks O2-induced airway hyper-reactivity, airway smooth muscle remodeling and mast cell accumulation in newborn rat lung, Pediatric Academic Societies, May 5–8, 2007, Toronto, ON, Abstract 4850.6).
METHODS
Materials.
Time-pregnant Sprague-Dawley rats were obtained from Charles River Laboratories (Raleigh, NC). Reagents were from Sigma Chemical Co., Aldrich (St. Louis, MO), unless otherwise stated. Antibodies to rat mast cell protease 1 (RMCP1, EC 3.4.21.39) and 2 were from Moredun Scientific (Midlothian, Scotland, UK). Secondary antibodies, avidin-biotin-peroxidase complex (ABC Elite), avidin-biotin blocking reagents, and diaminobenzidine substrate were obtained from Vector Laboratories (Burlingame, CA). Imatinib (Gleevec) was a gift from Novartis Pharmaceuticals, Basel, Switzerland. Cromolyn was obtained from ICN Pharmaceuticals, Costa Mesa, CA.
Animal exposures.
Procedures were approved by the Duke University Institutional Animal Care and Use committee. Newborn rats were exposed to 60% O2 or air alone beginning on the day of birth, as previously described (9). Rat pups from four litters were randomly sorted into four recombined litters and were exposed to 60% O2 or air in plastic cages fitted with sealed Plexiglas lids with adaptors for gas supply and venting as previously described in detail (11). The FiO2 was measured and recorded in each cage every 15 min with an analyzer equipped with a strip-chart recorder (model 572; Servomex, Norwood, MA). Humidity was routinely 70%. Nursing rat dams were switched between air and 60% O2 for every 24 h. Exposure to ambient air was <10 min/d for weighing and cage cleaning. Two litters were exposed in each treatment condition.
Cromolyn treatment.
Cromolyn is a mast cell stabilizer that prevents mast cell degranulation. Air and hyperoxia-exposed pups were injected intraperitoneally in the iliac fossa once daily with cromolyn 5 mg/kg/d or normal saline vehicle postnatal d 1 to 14.
Imatinib treatment.
Imatinib is a tyrosine-kinase inhibitor that targets the receptors for stem cell factor (c-kit), a regulator of mast cell homing (10). Air and hyperoxia-exposed pups were injected intraperitoneally with imatinib mesylate 50 mg/kg/d once daily or normal saline vehicle on postnatal d 5 to 14. Because imatinib also inhibits the PDGF receptor, we began treatment on postnatal d 5 in an effort to minimize potentially confounding effects of blocking PDGF actions on alveolar development and lung physiology (12).
Lung mechanics.
On 14th d of life, 6 to 8 pups/treatment group were anesthetized with ketamine 40 mg/kg and xylazine 10 mg/kg, tracheostomized with 20-GA tracheal cannulae, attached to an ECG monitor, and then attached to a mechanical ventilator equipped with pressure transducers (FlexiVent, SCIREQ, Montreal, PQ, Canada) (13). Anesthetized pups were given pancuronium bromide (0.8 mg/kg, i.p.) to block spontaneous breathing. We have previously described the methods in detail (14). A positive end-expiratory pressure (PEEP) of three was established by submerging the expiratory limb 3 cm below the surface of a water-filled graduated cylinder. After recruitment inflations, and after baseline airway resistance was stabilized, three increasing doses of methacholine (10, 25, and 100 mg/ml) were administered by nebulizer (Aeroneb, SCIREQ, Aerogen, Galway, Ireland) according to the manufacturer's directions, allowing an interval of ∼2 min between doses. Lungs were inflated to total lung capacity after each methacholine dose, which resulted in returning the airway resistance measurements back to baseline before subsequent doses. Total respiratory system resistance (RRS) was determined immediately before methacholine nebulization, and respiratory system compliance was measured as we have previously described in detail (15). Pups were euthanized with sodium pentobarbital 200 mg/kg i.p.
Mast cell abundance and degranulation.
After euthanasia, lungs were inflation fixed as described with 4% paraformaldehyde at 30 cm H2O pressure for ∼1 h, then immersion fixed overnight at 4°C. After fixation, tracheae were cut transversely into four cylindrical sections before embedding and sectioning. The right middle lobe of the lung was removed and cut perpendicular to the hilar bronchus to expose the major airways before paraffin embedding. Trachea and hilar airway sections were cut at 4 to 6 μm. Rehydrated sections were treated with 0.1% toluidine blue, 7% ethanol, and 1% sodium chloride (pH of 2.0–2.5) for 5 min to identify mast cells and mast cell granules, as previously described (16).
Morphometric measurements were performed on the right middle lobe because of previously described regional variations in lung maturation (17). Toluidine-stained sections of trachea (6 animals/treatment group, 5 sections/animal) and hilar bronchi (6 animals/treatment group, 5 sections/animal, and 4 random regions/section) were evaluated microscopically at 400× magnification. Total mast cell number in the trachea or randomly selected airway was normalized to basement membrane perimeter length, evaluated by digital image analysis software, and calibrated with a stage micrometer (Metamorph; Molecular Devices, West Chester, PA). Mast cells were categorized as intact, all granules were inside cell membrane, or degranulating, granules seen outside cell membrane. The proportion of degranulating mast cells were calculated: degranulating mast cells/total number of mast cells.
Separate sections were incubated in anti-RMCP 1 or 2. After dewaxing and quenching peroxidases in 0.3% H2O2 in methanol for 30 min, sections were permeabilized by trypsin digest (0.1% trypsin in 2% Tris, 0.1% calcium chloride, and distilled water) for 30 min. Sections were blocked with 5% horse serum then incubated with anti-RMCP 1 or 2, 1:250 for 1 h. RMCP 1 is found in connective tissue-type mast cells, and RMCP 2 is found in mucosal-type mast cells (18,19). Endogenous biotin was blocked using an avidin-biotin blocking kit. Sections were incubated in biotinylated horse anti-goat for RMCP 1, or horse anti-mouse for RMCP 2, 1:1000 for 1 h., followed by ABC and diaminobenzidine chromogen. RMCP 1 or 2 positive cells were evaluated microscopically at 400× magnification. These sections were qualitatively compared with toluidine sections.
Data analysis.
Data are presented as mean ± SEM. Group differences were evaluated with one-way ANOVA, followed by post hoc analysis using Tukey HSD (Kaleidagraph 3.6; Synergy Software, Reading, PA). Statistical significance was defined as a p < 0.05.
RESULTS
Body weight.
As expected, hyperoxia exposure impaired weight gain for all treatment groups, with a further small but nonsignificant decrement in oxy + imatinib treated pups (Fig. 1).

Effect of hyperoxia ± cromolyn or imatinib on body weight at P14. Data are expressed as mean ± SEM (n = 6/group). *p < 0.01 air vs oxy-exposed groups.
Respiratory system mechanics.
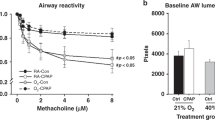
Hyperoxia + vehicle significantly increased methacholine-challenge induced total RRS at 100 mg/mL compared with untreated air-exposed pups (Fig. 2). Imatinib or cromolyn significantly decreased airways resistance in hyperoxia-exposed animals compared with the hyperoxia + vehicle group. The central airway resistance (Rn) contribution to total resistance was measured using a higher oscillation frequency, but its contribution to total resistance was small. Methacholine challenge did not affect central airways resistance significantly (data not shown). As expected, oxygen exposure impaired respiratory system compliance for all treatment groups. Imatinib also further impaired compliance in air and hyperoxia-exposed pups. Cromolyn treatment impaired compliance in the air-treated pups.

(A) RRS in cromolyn and imatinib treatment groups compared with air and oxy + vehicle measured by forced oscillometry. □ air + cromolyn; ○ air + vehicle; ⋄ oxy + vehicle; ▴ oxy + cromolyn; ♦ air + imtinib; and ▪ oxy + imatinib. Data are means of six per group ± SEM. *p < 0.05 vs oxy + vehicle. B, Respiratory system compliance (B) effects. Data are expressed as mean ± SEM. *p < 0.05 vs air + vehicle and **p < 0.05 vs oxy + vehicle.
Mast cell quantification and degranulation.
Toluidine-stained intact (Fig. 3C) and degranulating (Fig. 3D) mast cells were predominantly RMCP 1 positive (>95%) in all treatment conditions (Fig. 3E). They were located in the tracheal lamina propria and submucosa as well as the peribronchial adventitia and interstitium. Mast cell accumulation and degranulation were measured in the trachea and hilar airways. Cromolyn did not prevent tracheal mast cell accumulation in hyperoxia-exposed animals (Fig. 4A) but did reduce mast cell degranulation compared with the hyperoxia + vehicle group (Fig. 4B). There was a trend toward oxygen-induced mast cell accumulation in hilar airways. Cromolyn treatment reduced mast cell accumulation in hilar airways from oxygen but not air-exposed pups (Fig. 4C). Imatinib treatment totally prevented airway mast cell accumulation, whether determined by toluidine blue staining (Fig. 4A, C) or immunostaining with anti-RMCP 1 or RMCP 2, so degranulation ratios could not be assessed.

Mast cells (arrows) stained with toludine blue in hilar airway in (A) air and (B) hyperoxia-exposed newborn rat (200 ×, bar = 50 μm). (C) Intact mast cell and (D) in the initial stage of degranulation. (E) Anti-RMCP-1 labeled, connective tissue-type mast cells in trachea, and (F) nonimmune IgG labeled negative control (C–F, 1,000 ×, bar = 10 μm).

Mast cell accumulation and degranulation in the tracheal (A, B), and hilar (C, D) airways. Degranulation ratios were not done (ND) in imatinib-treated groups, because mast cell accumulation was absent. Data are expressed as means (n = 6 per group) ± SEM. *p < 0.05 vs. oxy + vehicle.
DISCUSSION
We found that hyperoxia-induced airway mast cell accumulation and methacholine-challenge-induced AHR in a newborn rat model of BPD characterized by heterogeneous alveolar hypoplasia and parenchymal thickening (9) that mimics aspects of human BPD (20,21). In support of our hypothesis that mast cells are essential to AHR development in a newborn model of BPD, we found that blocking mast cell degranulation by treatment with cromolyn or mast cell homing by treatment with imatinib prevented hyperoxia-induced AHR.
The mechanisms by which oxidative stress induces AHR in postnatal rat lung are not precisely known. Studies in juvenile rats exposed to hyperoxia (FIO2 >0.95) for 8 d demonstrated increased ASM mitosis and thickness (22), which would be expected to confer increased airway resistance given similar stimuli, compared with airways possessing normal ASM thickness. ASM cell proliferation is increased by mast cell products tryptase and platelet-derived growth factor (23). Cromolyn treatment would be expected to inhibit release of these mitogens, but we did not rigorously examine effects on ASM structure.
We found that cromolyn treatment blocked hyperoxia-induced mast cell degranulation in the trachea, but instead of blocking peripheral degranulation, cromolyn treatment blocked hyperoxia-induced accumulation of mast cells in peripheral airways. Mast cells may release granule contents without altering the integrity of their granules (24,25). This process has been termed “intragranular activation” (26). Cromolyn may have stabilized mast cell mediator release in the peripheral airways without affecting apparent degranulation. Because mast cells can release mediators without degranulating, blocking mast cell accumulation may be a more effective strategy to prevent mast cell effects in peripheral tissue. Because we are identifying mast cells by methods aimed at mast cell granules, our methods are unable to distinguish a completely degranulated mast cell from other leukocytes.
Accordingly, we also treated air and hyperoxia-exposed pups with imatinib, a tyrosine-kinase inhibitor that targets the receptors for c-kit, a regulator of mast cell homing. We found that imatinib treatment completely eliminated airway mast cell accumulation. Imatinib also prevented methacholine challenge-induced AHR in hyperoxia-exposed pups. Mast cell mediators, such as histamine and leukotrienes C4 and D4, can directly stimulate ASM contraction resulting in AHR (27) Mast cells and nerves communicate bidirectionally, giving rise to neurogenic inflammation, nerve growth, and ASM hypercontractility (28,29), which would be interrupted by blocking mast cell homing to airways.
We nevertheless observed that imatinib-treated pups (air and hyperoxia-exposed) had qualitatively impaired alveolar development (data not shown). Imatinib treatment did not affect total or large airway resistance at baseline measured by forced oscillometry. Given the potential for loss of alveolar tethering to contribute to AHR, we would have expected that impairment of alveolar development, reflected by the changes in baseline respiratory system compliance would have increased the sensitivity to methacholine challenge (30), but this was not observed.
Evaluating the role of mast cell accumulation in hyperoxia-induced neonatal, AHR would ideally use the specific manipulation of mast cells. Unfortunately, there are no ligands or receptors of which we are aware that are unique to mast cells. Rodent strains with abnormalities in loci encoding c-kit show defective mast cell recruitment, but this pathway is also important to other cell lineages, and potential effects on lung function have not been directly addressed in these strains (31,32). A mast cell deficient rat strain has been developed, but it sustains a high neonatal mortality associated with severe anemia, and the mechanism of mast cell deficiency is undefined (33,34). Transgenic and knockout mice offer a wider choice of models with defective mast cell function or disruptions of c-kit, but mouse airway anatomy and physiology (35) differ from other experimental animals, particularly the airway branching pattern and ASM pattern (36) limiting its usefulness for the study of AHR and asthma in newborn animals.
We found that hyperoxia had no effect on the type of airway mast cells seen, connective tissue-type (RMCP 1 positive), or mucosal-type (RMCP 2 positive). Previous reports observed both types of mature mast cells containing sufficient histamine and heparin to be identified by toluidine blue staining (8,37). We confirmed that the connective tissue-type mast cell was the predominant mast cell type in tracheal and bronchial lamina propria and smooth muscle regardless of exposure or treatment in 2-wk-old rats. This is consistent with previous reports of connective tissue-type mast cells predominating in air or hyperoxia-exposed newborn rats (8,19). Although we found few mast cells in the peripheral lung in air or hyperoxia-exposed P14 rat pups, adult rats demonstrate a more robust peripheral mast cell accumulation in perivascular regions (38). We did not perform a detailed analysis of mast cell abundance in lung periphery, and it is possible that subtle treatment effects could have been undetected.
We chose to use forced oscillometry to measure RRS because of important limitations in noninvasive plethysmographic methods (39). The advantage of this method is the ability to vary frequency to delineate the contribution of large and small airway resistance to total airway resistance. We observed no significant contribution of large/central airways resistance to total RRS. A disadvantage is that anesthesia and muscle paralysis will impair neurologic input to the respiratory system and will necessarily require positive pressure to achieve the measurements, which is dissimilar to spontaneously breathing patients with BPD. On balance, we thought it was preferable to avoid the artifacts and lack of precision inherent in plethysmography in neonatal rats.
Because of the association of pulmonary mast cell recruitment with clinical and experimental BPD, inhaled cromolyn has been tested for its ability to prevent BPD in premature newborns undergoing mechanical ventilation (40). Although treatment reduced a number of inflammatory mediators in the tracheal aspirates of these subjects, it did not reduce the incidence of BPD, as defined at the time.
In summary, immediate postnatal hyperoxia exposure of newborn rats increases methacholine-induced AHR, predominantly from the smaller airways, which is mediated in part by mast cells. Treatment with systemic cromolyn to block mast cell degranulation decreased methacholine-induced AHR in hyperoxia-exposed newborn rats. Inhibition of postnatal mast cell homing with imatinib blocked hyperoxia-induced AHR. We conclude that mast cells play a central role in the development of hyperoxia-induced postnatal AHR. Treatments directed at reducing mast cell accumulation or degranulation may interrupt the development of AHR in patients developing BPD.
Abbreviations
- AHR:
-
airway hyper-reactivity
- ASM:
-
airway smooth muscle
- RMCP:
-
rat mast cell proteases
References
Kennedy JD, Edward LJ, Bates DJ, Martin AJ, Dip SN, Haslam RR, McPhee AJ, Staugas RE, Baghurst P 2000 Effects of birthweight and oxygen supplementation on lung function in late childhood in children of very low birth weight. Pediatr Pulmonol 30: 32–40
Halvorsen T, Skadberg BT, Eide GE, Roksund O, Aksnes L, Oymar K 2005 Characteristics of asthma and airway hyper-responsiveness after premature birth. Pediatr Allergy Immunol 16: 487–494
Martin RJ, Walsh-Sukys MC 1999 Bronchopulmonary dysplasia—no simple solution. N Engl J Med 340: 1036–1038
Baraldi E, Bonetto G, Zacchello F, Filippone M 2005 Low exhaled nitric oxide in school-age children with bronchopulmonary dysplasia and airflow limitation. Am J Respir Crit Care Med 171: 68–72
Brightling CE, Bradding P, Pavord ID, Wardlaw AJ 2003 New insights into the role of the mast cell in asthma. Clin Exp Allergy 33: 550–556
Subramaniam M, Sugiyama K, Coy DH, Kong Y, Miller YE, Weller PF, Wada K, Wada E, Sunday ME 2003 Bombesin-like peptides and mast cell responses: relevance to bronchopulmonary dysplasia?. Am J Respir Crit Care Med 168: 601–611
Lyle RE, Tryka AF, Griffin WS, Taylor BJ 1995 Tryptase immunoreactive mast cell hyperplasia in bronchopulmonary dysplasia. Pediatr Pulmonol 19: 336–343
Denis D, Fayon MJ, Berger P, Molimard M, De Lara MT, Roux E, Marthan R 2001 Prolonged moderate hyperoxia induces hyperresponsiveness and airway inflammation in newborn rats. Pediatr Res 50: 515–519
Yi M, Jankov RP, Belcastro R, Humes D, Copland I, Shek S, Sweezey NB, Post M, Albertine KH, Auten RL, Tanswell AK 2004 Opposing effects of 60% oxygen and neutrophil influx on alveologenesis in the neonatal rat. Am J Respir Crit Care Med 170: 1188–1196
Akin C, Brockow K, D'Ambrosio C, Kirshenbaum AS, Ma Y, Longley BJ, Metcalfe DD 2003 Effects of tyrosine kinase inhibitor STI571 on human mast cells bearing wild-type or mutated c-kit. Exp Hematol 31: 686–692
Han RN, Buch S, Tseu I, Young J, Christie NA, Frndova H, Lye SJ, Post M, Tanswell AK 1996 Changes in structure, mechanics, and insulin-like growth factor-related gene expression in the lungs of newborn rats exposed to air or 60% oxygen. Pediatr Res 39: 921–929
Bostrom H, Gritli-Linde A, Betsholtz C 2002 PDGF-A/PDGF alpha-receptor signaling is required for lung growth and the formation of alveoli but not for early lung branching morphogenesis. Dev Dyn 223: 155–162
Pillow JJ, Korfhagen TR, Ikegami M, Sly PD 2001 Overexpression of TGF-alpha increases lung tissue hysteresivity in transgenic mice. J Appl Physiol 91: 2730–2734
Kallapur SG, Moss TJ, Auten RL Jr, Nitsos I, Pillow JJ, Kramer BW, Maeda DY, Newnham JP, Ikegami M, Jobe AH 2009 IL-8 signaling does not mediate intra-amniotic LPS-induced inflammation and maturation in preterm fetal lamb lung. Am J Physiol Lung Cell Mol Physiol 297: L512–L519
Auten RL, Potts EN, Mason SN, Fischer B, Huang Y, Foster WM 2009 Maternal exposure to particulate matter increases postnatal ozone-induced airway hyperreactivity in juvenile mice. Am J Respir Crit Care Med 180: 1218–1226
Baud O, Zupan V, Lacaze-Masmonteil T, Dehan M 1999 Neurological adverse effects of early postnatal dexamethasone in very preterm infants. Arch Dis Child Fetal Neonatal Ed 80: F159
Brumley GW, Chernick V, Hodson WA, Normand C, Fenner A, Avery ME 1967 Correlations of mechanical stability, morphology, pulmonary surfactant, and phospholipid content in the developing lamb lung. J Clin Invest 46: 863–873
Steptoe RJ, McMenamin PG, McMenamin C 1994 Distribution and characterisation of rat choroidal mast cells. Br J Ophthalmol 78: 211–218
Wilkes LK, McMenamin C, Holt PG 1992 Postnatal maturation of mast cell subpopulations in the rat respiratory tract. Immunology 75: 535–541
Bland RD 2005 Neonatal chronic lung disease in the post-surfactant era. Biol Neonate 88: 181–191
Jobe AJ 1999 The new BPD: an arrest of lung development. Pediatr Res 46: 641–643
Hershenson MB, Kelleher MD, Naureckas ET, Abe MK, Rubinstein VJ, Zimmermann A, Bendele AM, McNulty JA, Panettieri RA, Solway J 1994 Hyperoxia increases airway cell S-phase traversal in immature rats in vivo. Am J Respir Cell Mol Biol 11: 296–303
Page S, Ammit AJ, Black JL, Armour CL 2001 Human mast cell and airway smooth muscle cell interactions: implications for asthma. Am J Physiol Lung Cell Mol Physiol 281: L1313–L1323
Paus R, Theoharides TC, Arck PC 2006 Neuroimmunoendocrine circuitry of the ‘brain-skin connection.'. Trends Immunol 27: 32–39
Slominski AT 2006 Proopiomelanocortin signaling system is operating in mast cells. J Invest Dermatol 126: 1934–1936
Theoharides TC, Cochrane DE 2004 Critical role of mast cells in inflammatory diseases and the effect of acute stress. J Neuroimmunol 146: 1–12
Bradding P, Walls AF, Holgate ST 2006 The role of the mast cell in the pathophysiology of asthma. J Allergy Clin Immunol 117: 1277–1284
Bauer O, Razin E 2000 Mast cell-nerve interactions. News Physiol Sci 15: 213–218
Piedimonte G 2003 Contribution of neuroimmune mechanisms to airway inflammation and remodeling during and after respiratory syncytial virus infection. Pediatr Infect Dis J 22: S66–S74; discussion S74–S65
Mortola JP 1983 Dysanaptic lung growth: an experimental and allometric approach. J Appl Physiol 54: 1236–1241
Lee DM, Friend DS, Gurish MF, Benoist C, Mathis D, Brenner MB 2002 Mast cells: a cellular link between autoantibodies and inflammatory arthritis. Science 297: 1689–1692
Zhou JS, Xing W, Friend DS, Austen KF, Katz HR 2007 Mast cell deficiency in Kit(W-sh) mice does not impair antibody-mediated arthritis. J Exp Med 204: 2797–2802
Liu S, Chihara K, Maeyama K 2005 The contribution of mast cells to the late-phase of allergic asthma in rats. Inflamm Res 54: 221–228
Niwa Y, Kasugai T, Ohno K, Morimoto M, Yamazaki M, Dohmae K, Nishimune Y, Kondo K, Kitamura Y 1991 Anemia and mast cell depletion in mutant rats that are homozygous at “white spotting (Ws)” locus. Blood 78: 1936–1941
Irvin CG, Bates JH 2003 Measuring the lung function in the mouse: the challenge of size. Respir Res 4: 4
Wenzel S, Holgate ST 2006 The mouse trap: It still yields few answers in asthma. Am J Respir Crit Care Med 174: 1173–1176 discussion 1176–1178.
McMenamin PG, Morrison SM, McMenamin C 1996 Immunomorphologic studies of mast cell heterogeneity, location, and distribution in the rat conjunctiva. J Allergy Clin Immunol 97: 1375–1386
Brock TG, Di Giulio C 2006 Prolonged exposure to hyperoxia increases perivascular mast cells in rat lungs. J Histochem Cytochem 54: 1239–1246
Bates J, Irvin C, Brusasco V, Drazen J, Fredberg J, Loring S, Eidelman D, Ludwig M, Macklem P, Martin J, Milic-Emili J, Hantos Z, Hyatt R, Lai-Fook S, Leff A, Solway J, Lutchen K, Suki B, Mitzner W, Pare P, Pride N, Sly P 2004 The use and misuse of Penh in animal models of lung disease. Am J Respir Cell Mol Biol 31: 373–374
Viscardi RM, Hasday JD, Gumpper KF, Taciak V, Campbell AB, Palmer TW 1997 Cromolyn sodium prophylaxis inhibits pulmonary proinflammatory cytokines in infants at high risk for bronchopulmonary dysplasia. Am J Respir Crit Care Med 156: 1523–1529
Acknowledgements
We thank Mary Whorton for the technical assistance. We also thank Drs. David Tanaka, Mary Sunday, and Ronald Goldberg for the helpful discussions.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supported by Children's Miracle Network, grant HD-043728 from the National Institutes of Child Health and Human Development, grant HL-067021 from the National Heart, Lung and Blood Institute, and grants ES-011961 and -012496 from the National Institute of Environmental Health Sciences.
Rights and permissions
About this article
Cite this article
Schultz, E., Potts, E., Mason, S. et al. Mast Cells Mediate Hyperoxia-Induced Airway Hyper-Reactivity in Newborn Rats. Pediatr Res 68, 70–74 (2010). https://doi.org/10.1203/PDR.0b013e3181e0cd97
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/PDR.0b013e3181e0cd97
This article is cited by
-
Intra-amniotic LPS amplifies hyperoxia-induced airway hyperreactivity in neonatal rats
Pediatric Research (2013)
