
Abstract
Premature infants are at risk for bilirubin-associated brain damage. In cell cultures bilirubin causes neuronal apoptosis and necrosis. Ibuprofen is used to close the ductus arteriosus, and is often given when hyperbilirubinemia is at its maximum. Ibuprofen is known to interfere with bilirubin–albumin binding. We hypothesized that bilirubin toxicity to cultured rat embryonic cortical neurons is augmented by coincubation with ibuprofen. Incubation with ibuprofen above a concentration of 125 μg/mL reduced cell viability, measured by methylthiazole tetrazolium reduction, to 68% of controls (p < 0.05). Lactate dehydrogenase (LDH) release increased from 29 to 38% (p < 0.01). The vehicle solution did not affect cell viability. Coincubation with 10 μM unconjugated bilirubin (UCB)/human serum albumin in a molar ratio of 3:1 and 250 μg/mL ibuprofen caused additional loss of cell viability and increased LDH release (p < 0.01), DNA fragmentation, and activated caspase-3. Preincubation with the pan-caspase inhibitor z-val-ala-asp-fluoromethyl ketone abolished ibuprofen- and UCB-induced DNA fragmentation. The study demonstrates that bilirubin in low concentration of 10 μM reduces neuron viability and ibuprofen increases this effect. Apoptosis is the underlying cell death mechanism.
Similar content being viewed by others

Main
Serum bilirubin levels above 430 μM have been shown to cause bilirubin encephalopathy and kernicterus in term infants (1). Previous studies revealed occurrence of kernicterus in very low birth weight infants with relation to low serum bilirubin levels (2–5). Even though kernicterus is rare in premature infants, there is a risk of bilirubin-induced brain injury (6). After adjustment for various demographic and clinical risk factors, Oh et al. (6) found a significant association between peak serum bilirubin (140 ± 43 μM) and death or neurodevelopmental impairment, psychomotor developmental index <70, and hearing impairment in extremely low birth weight infants (gestational age 26.2 ± 2.1 wk; birth weight 783 ± 139 g).
Ibuprofen has been proposed for closing the ductus arteriosus alternatively to indomethacin (7). Ductus interventions are frequently performed at an age when hyperbilirubinemia is present. As ibuprofen interferes with bilirubin–albumin binding it may increase the risk of bilirubin-associated encephalopathy (8). Mathematical models for brain bilirubin uptake as a function of plasma total bilirubin concentration and bilirubin–albumin dissociation rate constant exist (9,10). Ahlfors and Parker (11) suggested that plasma-free bilirubin concentration provides rates of brain bilirubin uptake consistent with the reported bilirubin production rate and miscible bilirubin pool distribution. Clinical studies on bilirubin neurotoxicity point in the same direction as abnormal auditory brainstem responses in jaundiced newborns increased with increasing unbound bilirubin concentrations but not with increasing total bilirubin concentrations (12). At ibuprofen concentrations of 200 μg/mL, the free fraction of bilirubin increased by a factor of 3.9 in a sample of adult and newborn infant serum with a bilirubin/albumin molar ratio of 0.5 (13). Ahlfors (8) observed a twofold increase at an ibuprofen level of 200 μg/mL. Clinical relevance of ibuprofen–albumin–bilirubin interactions in preterm infants after treatment remains to be determined (Arnada JV, et al. Plasma Unbound Bilirubin in Placebo and Ibuprofen Treated Preterm Neonates, PAS 2008 Annual Meeting, May 3–6, 2009, Honolulu, Hawaii, Abstract 5844.9). Sulfisoxazole or ceftriaxone interacting with bilirubin–albumin binding increased the bilirubin concentration as well (8). These results suggest that ibuprofen may increase the risk of bilirubin encephalopathy in jaundiced newborn infants. Studies on neuronal and astrocytic cultures showed apoptosis and necrosis induced by unconjugated bilirubin (UCB) at varied molar ratios of bilirubin/albumin (14,15). One study tested the hypothesis that free bilirubin measured in culture medium, rather than total UCB predicts the toxicity of bilirubin in several cell lines (16). Neuronal damage by ibuprofen itself was not reported. We hypothesized that ibuprofen augments UCB toxicity to cortical neurons.
The aim of the study was to investigate the effects of UCB in combination with ibuprofen in primary neuronal cultures.
METHODS
Primary cortical neuronal culture.
Primary neuronal cultures were prepared from cortices of Wistar rat embryos (E16–E18) (Forschungseinrichtung für Experimentelle Medizin, Charité–Universitätsmedizin Berlin, Germany) as previously described (17) with minor modifications. All procedures were performed according to the local guidelines for animal research approved by the Charité, Universitätsmedizin Berlin. In brief, cerebral cortices were dissected in serum-free neurobasal medium with B27 supplement (GIBCO, Invitrogen GmbH, Karlsruhe, Germany), meninges were removed, and tissue was incubated for 15 min in trypsin/ethylenediaminetetraacetic acid (0.05/0.02% wt/vol in phosphate-buffered saline [PBS]) at 37°C, rinsed twice with PBS and once with dissociation medium (neurobasal medium with 2% B27 supplement, 1% penicillin, and streptomycin and 1% l-glutamine), mechanically dissociated by repeated aspirations through a Pasteur pipette in dissociation medium, pelleted by mild centrifugation (1200 U/min, 2 min at room temperature), and seeded in flat-bottom wells in serum-free neurobasal medium with B27 supplement containing 2 mM l-glutamine, at a density of 3 × 106/mL. Before cell plating, wells were treated with poly-d-lysine (0.5% wt/vol in aqua; Sigma Chemical, St. Louis, MO) for 1 h at room temperature and rinsed twice with aqua. Cells were kept in a fully humidified incubator at 37°C with 5% CO2 and fed every 3–4 d. Neurons were characterized by indirect immunocytochemistry for neurofilament. A purity of 99.5% neuronal cells was reached.
Cell treatment with unconjugated bilirubin or ibuprofen.
After a total of 14–16 d in vitro, cells were exposed to ibuprofen (7.5–1000 μg/mL) or 1 and 10 μM UCB/human serum albumin (HSA) alone for 48 h. Limited to our study model we simultaneously coincubated UCB 1 or 10 μM with 125 or 250 μg/mL ibuprofen. Control cells remained untreated. UCB (Sigma Chemical) was dissolved freshly in 0.1 M NaOH and used at a UCB/HSA molar ratio of 3:1. We used dissolved ibuprofen-THAM (Pedea, Orphan Europe, Paris, France). To exclude the effect of the dissolving buffer, we incubated the cells in corresponding concentrations. Experiments with UCB were performed in the dark to minimize photoisomerization. To achieve interaction of UCB with the cells a UCB/HSA ratio of 3:1 was used, mimicking the toxic conditions of prolonged hyperbilirubinemia (18). For the same reason, we used HSA because the cellular uptake of bilirubin bound to HSA is more rapid than for bovine serum albumin-bound bilirubin (19).
To explore the cellular mechanisms by which bilirubin or ibuprofen can affect neuronal cells, cultures were additionally treated, in some experiments, with the protein kinase inhibitor staurosporine (Sigma Chemical) at a concentration 500 nm, an agent that induces apoptosis (20) or the pan-caspase inhibitor z-val-ala-asp-fluoromethyl ketone (zVAD-fmk) (Promega, Mannheim, Germany) (20 μM). Staurosporine was solved in dimethylsulfoxide (DMSO) with a final DMSO concentration of 0.05%.
Cell viability assay.
The tetrazolium salt assay relies on the reduction of methylthiazole tetrazolium (MTT) (3-(4,5-dimethylthiazole-2-yl)-2,5-diphenyltetrazoliumbromide) (Sigma Chemical) to colored formazan by active dehydrogenases in live mitochondria to estimate the number of viable cells (21). Neuronal cells were plated onto 96 plates well coated with poly-d-lysine in 50 μL aliquots (60,000 cells/well) and treated with UCB/HSA (3:1), ibuprofen, or a combination of both. Control cells were kept in neurobasal medium. At the end of the experiment, 5 μL MTT at a final concentration of 0.5 mg/mL was added for 2 h. Violet formazan dye grain was solubilized by adding 100 μL of 10% sodium dodecyl sulfate (SDS)—0.01 N HCl. Absorbance at 570 nM and a reference wavelength of 630 nm were read using a microplate reader (BioRad, Munich, Germany).
Cell death analysis by lactate dehydrogenase release assay.
Cell injury was assessed by determining lactate dehydrogenase (LDH) release in the cell incubating medium of cultured cells, using a Roche Cytotoxicity Detection Kit (Roche Diagnostics, Mannheim, Germany, Cat. No. 1644793). Absorbance at 490 nM and 630 nm was read using a microplate reader (BioRad, Munich, Germany). The reference wavelength of 630 nm was used according to the recommendations in the kits manual. Cell total LDH was obtained after exposure of cultures to 2% Triton-X-100 (Sigma Chemical). LDH release was expressed as a percentage of LDH in medium to total LDH.
Light phase contrast photomicrographs.
Cells were exposed to UCB/HSA 10 μM, ibuprofen 250 μg/mL, or both. Control cells were kept in neurobasal medium. Photomicrographs were taken with an Olympus digital camera.
Western blot activated caspase-3.
After incubation for 6 h, cells were harvested and washed once with PBS. The pellet was homogenized in RIPA buffer (1% NP40, 0.5% sodium deoxycholate, 0.1% SDS, 1 mM EDTA, 1 mM EGTA, 1 mM Na3VO4, 20 mM NaF, 0.5 mM DTT, 1 mM phenylmethyl sulfonyl fluoride, and protease inhibitor cocktail in PBS pH 7.4) and phenylmethyl sulfonyl fluoride as protease inhibitor in PBS pH 7.4. The homogenate was centrifuged at 1050g (4°C) for 10 min, and the microsomal fraction was subsequently centrifuged at 17,000g (4°C) for 20 min. Twenty micrograms of the resulting cytosolic protein extracts were heat denaturated in Laemmli sample loading buffer, separated by 10% polyacrylamide gel electrophoresis, and electrotransferred onto a nitrocellulose membrane. Nonspecific protein binding was prevented by treating the membrane with 5% nonfat dry milk in Tris-buffered saline/0.1% Tween 20 for 2 h at room temperature. Next, the membrane was incubated overnight at 4°C with primary rabbit polyclonal cleaved caspase-3 antibody (1:2000; Cell Signaling, Danvers, MA). Secondary incubations were performed with horseradish peroxidase-linked anti-rabbit antibody (1:8000; Cell Signaling). Bands were visualized using enhanced chemiluminescence (Cell Signaling), and serial exposures were made to radiographic film (Hyperfilm enhanced chemiluminescence; Amersham Pharmacia Biosciences, Buckinghamshire, UK). Densitometric analysis of the blots was performed with an image analysis program (BioDocAnalyze, Whatman Biometra, Goettingen, Germany). For stripping, membranes were incubated with stripping buffer (100 mM β-mercaptoethanol, 2% SDS, 62.5 mM Tris-HCl, pH 6.7) at 50°C for 30 min, then washed, blocked, and reincubated overnight at 4°C with mouse anti-β-actin MAb (1:5000; Sigma Chemical) and with the secondary anti-mouse antibody (1:10000; Dako, Glostrup, Denmark) for 2 h at RT.
Apoptosis detection enzyme-linked immunosorbent assay.
To quantify apoptosis, histone-complexed DNA fragments from the cytoplasm of apoptotic cells were detected immunochemically and the amount determined with spectrophotometry. After incubation of cells with UCB/HSA (3:1) or ibuprofen for 6 h as described, cell incubating medium, which may have contained necrotic DNA, was discarded. Apoptotic cells were lysed and isolated by centrifugation and the amount of apoptotic nucleosomes in the cell lysate was determined using the Cell Death Detection enzyme-linked immunosorbent assay (Roche Diagnostics, Cat No. 11 920685 001). As a positive control for detection of apoptotic cell death, cells were incubated with the protein kinase inhibitor staurosporine (1 mM stock solution dissolved in DMSO) and were used at a final concentration of 500 nM. To inhibit apoptotic cell death, the pan-caspase inhibitor zVAD-fmk was used in a final concentration of 20 μM.
Statistics.
Data were analyzed using GraphPad Prism 4 for Windows (GraphPad Software, San Diego, CA). Cells from at least six wells from six separate experiments were examined for all experimental conditions. Data are given as mean ± SEM. Statistical significance was evaluated with the one-way analysis of variance with Dunnett's multiple comparison test unless otherwise indicated. A p value of <0.05 was considered significant.
RESULTS
Ibuprofen reduces cell viability.
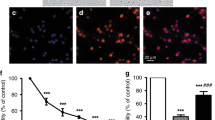
To determine the influence of increasing concentrations of ibuprofen alone on neuronal survival, we evaluated the cell viability by MTT reduction. After incubation of ibuprofen alone in concentrations ranging from 7.5 to 1000 μg/mL for 48 h, cell viability (Fig. 1) was reduced significantly in concentrations of above 125 μg/mL (68% of controls). Cell death, measured by LDH release increased likewise from 29% in controls to 38% at 250 μg/mL ibuprofen. Ibuprofen was dissolved in trometamol buffer. Incubating the cells with trometamol buffer (vehicle) in concentrations of that of the vial containing ibuprofen did not affect the cells (data not shown).

MTT reduction (white columns) in cells exposed to different ibuprofen concentrations for 48 h. Above a concentration of 125 μg/mL MTT reduction was decreased. LDH release (gray columns), demonstrated as percentage of total, increased above the same concentration. Con = controls without ibuprofen (data are mean ± SEM, n = 6, **p < 0.01).
Unconjugated bilirubin in association with ibuprofen reduces cell viability.
To determine the combined effect of UCB/HSA (3:1) and ibuprofen, we incubated the cultures for 48 h with both substances. Light phase contrast photographs showed slightly reduced viability of neurons after incubation with 10 μM UCB/HSA (3:1) or 250 μg/mL ibuprofen alone compared with controls. Cells incubated with both substances were markedly affected (Fig. 2A). UCB/HSA (3:1) concentration of 1 μM alone or in combination with ibuprofen (125 or 250 μg/mL) did not reduce cell viability nor increase LDH release significantly (Fig. 2B). In the case of incubation with 10 μM UCB/HSA (3:1), the MTT assay revealed reduced cell viability of 48%. No significant increase in the release of LDH was detected. By coincubation with 250 μg/mL ibuprofen, cell viability was reduced dramatically (2% of controls) and LDH release increased from 36 to 56%.

(A) Typical transmission light phase contrast photomicrographs of primary neuronal cultures after 48 h of incubation with medium (control), UCB/HSA (10 μM), ibuprofen (ibu) (250 μg/mL), or both substances (UCB/HSA + ibu). Cells exposed to UCB/HSA or ibuprofen alone exhibited morphologic features of apoptosis with larger nuclei and shorter cell processes. Incubation with UCB/HSA and ibuprofen damaged the cells severely: cell bodies and processes were nearly lost. Magnification 40×, scale bar = 100 μm. (B) MTT reduction in percent of controls (white columns) after treatment with unconjugated bilirubin (1 and 10 μM) in combination with ibuprofen (125 and 250 μg/mL) for 48 h. Coincubation of UCB/HSA 10 μM and ibuprofen 250 μg/mL reduced MTT cleavage. LDH release (gray columns) increased after treatment with UCB/HSA 10 μM in combination with ibuprofen 250 μg/mL (Data are mean ± SEM, n = 6, **p < 0.01).
UCB/HSA and ibuprofen induce apoptosis.
To further specify the type of cell death, we assessed protein samples obtained from treated cells by Western blot analysis for activation of caspase-3 which plays a key role in the apoptotic pathway by initiating DNA fragmentation (Fig. 3). The analysis indicated an increase of activated caspase-3 for neuronal cells incubated with 250 μg/mL ibuprofen or coincubated cells. The increase by incubation with 10 μM UCB/HSA (3:1) alone was not significant.

(A) Western blot densitometric quantification of protein levels for cleaved caspase-3 increased after treatment with UCB/HSA (10 μM) (not significant), ibuprofen (250 μg/mL), and coincubation. Columns represent the density ratios of cleaved caspase-3 to β-actin in percent (data are mean ± SEM, n = 9, **p < 0.01, §p < 0.001) Kruskal-Wallis test with Dunn's multiple comparison test. (B) Representative blot out of a series of nine blots from three experiments.
Analyzing the amount of oligonucleosomal DNA fragments indicating apoptosis revealed an increase for neurons incubated with 250 μg/mL ibuprofen (140% of controls) and for coincubated cells (178% of controls) (Fig. 4). Staurosporine increased the amount to 220% of controls.

Oligosomal DNA content in percent of controls as indicator for apoptosis. UCB/HSA (10 μM) coincubated with ibuprofen 250 μg/mL increased oligosomal DNA compared with controls. Ibuprofen alone also increased it, whereas vehicle treatment did not damage cells. Staurosporine (SSP) (500 nM) increased oligosomal DNA (data are mean ± SEM, n = 6, *p < 0.05, **p < 0.01).
Coincubation-induced apoptosis is attenuated by the pan-caspase inhibitor zVAD-fmk.
UCB/HSA (10 μM) and ibuprofen (250 μg/mL) augmented the amount of oligonucleosomal DNA compared with controls. In cells pretreated with zVAD-fmk (20 μM), apoptosis could be blocked. Treatment with zVAD-fmk reduced oligosomal DNA also in control cells (Fig. 5).

Oligosomal DNA content in percent of controls as indicator for apoptosis. Coincubation of ibuprofen (250 μg/mL) and UCB/HSA (10 μM) increased oligosomal DNA compared with controls. zVAD-fmk (20 μM) blocked coincubation-induced apoptosis. Staurosporine (SSP) increased the amount of apoptotic DNA as well. ZVAD-fmk alone decreased the amount of oligosomal DNA significantly (data are mean ± SEM, n = 6, **p < 0.01).
DISCUSSION
The main finding of the present study is that ibuprofen not only caused neuronal damage in increasing concentrations but also it augmented bilirubin toxicity in rat embryonic neuronal cortical culture. Toxicity of UCB to neural cells has been demonstrated in vivo (22,23) and in vitro, mechanisms and reasons for cell-specific damage remain unclear (14,15,24,25). An advantage of primary neuronal culture is the high survival of plated cells in serum-free B27/neurobasal medium (17).
In the present study, bilirubin concentrations (1–10 μM UCB/HSA at a molar ratio of 3:1) were low to moderate compared with in vivo concentrations of 100 μM or even higher in preterm infants, but in higher concentrations UCB causes excessive damage in vitro (14,15). In different brain cells calculated free bilirubin concentrations in the range of 0.071–0.77 μM induced neurotoxicity in vitro (26). In one study, these levels of free bilirubin concentrations could be calculated in vivo (27). A limitation of our study is that we did not measure free bilirubin concentrations. Studies on bilirubin displacement from albumin binding showed increasing free bilirubin concentrations with rising ibuprofen concentration (8,13). Most in vitro studies on bilirubin toxicity correlated cell damage to unbound bilirubin concentrations (14,15). Only one study analyzed the toxicity of free bilirubin measured in culture medium, rather than total UCB in several cell lines (16). A clinical study measuring auditory brainstem responses in jaundiced newborns found abnormal responses with increasing unbound bilirubin concentrations but not with increasing total bilirubin concentrations (12).
Our ibuprofen concentrations of 30–250 μg/mL corresponded to concentrations in treated infants. After infusion of 10 mg/kg ibuprofen-lysine within the first 3 h after birth, plasma concentration at 1 h was 180.6 ± 11.1 mg/L with a range between 107 and 280.5 mg/L (28). Plasma concentrations for ibuprofen of 43. 5 ± 11.2 mg/L were reported with the same dose on day 3 of life (29). With 20 mg/kg median concentrations reached 68 μg/mL (30). Plasma half-life was prolonged 10 times in preterm infants compared with infants of more than 3 mo of age (28).
Even if the ibuprofen concentrations used in our models are high, the finding that ibuprofen caused independent neurotoxicity above a concentration of 125 μg/mL is of greatest concern especially when ibuprofen displaces bilirubin from the albumin binding and causes additional cell damage.
Treatment with a combination of ibuprofen and UCB/HSA (molar ration 3:1) showed dramatically reduced cell viability. Protein binding of ibuprofen was found between 75 (13) and 95% (28). Ibuprofen interfered with bilirubin–albumin binding and increased the amount of unbound bilirubin. Above a concentration of 50 μg/mL ibuprofen UCB was significantly increased and the impact on bilirubin–albumin binding greatly exceeded that of sulfisoxazole (8). As UCB causes neuronal damage in vivo and in vitro, the additional effect of ibuprofen on bilirubin toxicity is caused by higher UCB levels. Coincubation of the cells with 10 μM UCB/HSA (3:1) and 250 μg/mL ibuprofen, as used in our model, appears to represent the most toxic event.
Ibuprofen has been shown to induce apoptosis in the murine microglial cell line BV-2 (31). Cell viability was reduced at 250 μM (≅500 μg/mL) whereas we found reduced viability in cortical neuronal culture already above 125 μg/mL.
In another model of ischemia-induced neuronal damage, ibuprofen showed neuroprotective effects in rat forebrain and in neuronal/microglial cocultures and no neurotoxic effect in concentrations of 500 μM (=1000 μg/mL) (32). This difference to our result may be explained by the microglial apoptosis or different vulnerability in vivo and in vitro. Elimination of inflammatory cells as microglia and of their proinflammatory cytokines from the central nervous system by apoptosis plays an important role in protecting neurons (33). Other authors found neuroprotective effects in rat hippocampal neurons by ibuprofen against amyloid proteins (Aβs), a model for Alzheimer's disease, as well as Aβ-induced aggravation of glutamate toxicity (34). They did not find reduced viability by ibuprofen alone in a concentration of 100 μM (≅200 μg/mL). Other studies showed a protective effect of ibuprofen in Aβ-induced damage in vivo and in vitro. Ibuprofen seems to interact with several pathways, including the inhibition of cyclooxygenases (35) and production of nitric oxide (NO). Excessive NO release from activated microglia has a pivotal role in neuronal death. The underlying mechanism of ibuprofen has been shown to be the reduction of inducible nitric oxide synthesis in different cell lines (36). The neuroprotective effects of ibuprofen in all described models underlie the mechanism of eliminating inflammatory cells.
Coincubation of the cells with 10 μM UCB/HSA (3:1) and 250 μg/mL ibuprofen showed reduced cell viability measured by MTT and LDH assay as demonstrated in Figure 2. Lack of increased LDH release for cells incubated with 10 μM UCB or 10 μM UCB and 125 μg/mL ibuprofen could be explained by the MTT assay being the more sensitive assay in detecting cytotoxic events compared with LDH leakage (37). For the same reason only coincubated cells showed increased DNA fragmentation (38). Furthermore, MTT and LDH assay did not detect cell damage of cells incubated with 1 μM UCB/HSA (3:1) and 250 μg/mL compared with cells incubated with 250 μg/mL ibuprofen alone. This effect may be explained by a neuroprotective action of bilirubin in low concentrations against oxidative stress (39). Elevated LDH release in control cells may be explained by replacing neurobasal medium and its effect on the treated cells. This could explain the absence of LDH release in bilirubin experiments. The findings in our experiments suggest evaluating mitochondrial dysfunction to understand differences between MTT reduction and LDH release. Further studies should investigate cytochrome c release from mitochondria in response to incubation with UCB (40).
Our study shows that the underlying mechanism of damage is apoptosis. Coincubating the cell with both substances increased the number of DNA fragments. In the human neuronal cell line NT-2, Hanko et al. (15) found apoptotic cell damage in low and moderate bilirubin concentrations ≤25 μM and necrosis in higher concentrations. The study by Grojean et al. (25) reported predominantly apoptotic cell death in low UCB concentrations whereas Silva et al. (41) induced apoptosis in fetal rat neurons in high UCB concentrations (17 and 86 μM).
As described by other authors, the pan-caspase inhibitor zVAD-fmk reduced neuronal apoptosis induced by UCB (24).
The strength of our study is the use of embryonic neuronal cultures and the approach used to detect the effect of UCB/HSA (3:1) and ibuprofen with different methods, MTT reduction and LDH release for cell viability, DNA fragmentation enzyme-linked immunosorbent assay and Western blot for apoptosis. On the other hand, the study is limited as an in vitro model. On the basis of this model, further investigations should be done including measurement of free bilirubin, bilirubin displacement, and involvement of mitochondrial dysfunction in bilirubin toxicity. Being cultivated for 14–16 d, the neurons were mature. Further data should be obtained treating immature neuronal cells after 4–5 d in culture and comparing them with the results in our study as other groups showed that UCB is more toxic in immature neuronal cells (40). We conclude that bilirubin is highly toxic to primary embryonic neuronal cultures in low concentration and that ibuprofen is toxic in therapeutic concentration. Combined exposure is more toxic than exposure to either substance alone. Both toxicities are due to augmented apoptosis and ibuprofen toxicity can be inhibited by zVAD-fmk. These findings may be important for the treatment of preterm infants with both jaundice and patent ductus arteriosus.
Abbreviations
- HSA:
-
human serum albumin
- LDH:
-
lactate dehydrogenase
- MTT:
-
3-(4,5-dimethylthiazole-2-yl)-2,5-diphenyltetrazoliumbromide
- UCB:
-
unconjugated bilirubin
- zVAD-fmk:
-
z-val-ala-asp-fluoromethyl ketone
References
Ebbesen F, Andersson C, Verder H, Grytter C, Pedersen-Bjergaard L, Petersen JR, Schaarup J 2005 Extreme hyperbilirubinaemia in term and near-term infants in Denmark. Acta Paediatr 94: 59–64
Stern L, Denton RL 1965 Kernicterus in small premature infants. Pediatrics 35: 483–485
Cashore WJ, Oh W 1982 Unbound bilirubin and kernicterus in low-birth-weight infants. Pediatrics 69: 481–485
Gartner LM, Snyder RN, Chabon RS, Bernstein J 1970 Kernicterus: high incidence in premature infants with low serum bilirubin concentrations. Pediatrics 45: 906–917
Harris RC, Lucey JF, Maclean JR 1958 Kernicterus in premature infants associated with low concentrations of bilirubin in the plasma. Pediatrics 21: 875–884
Oh W, Tyson JE, Fanaroff AA, Vohr BR, Perritt R, Stoll BJ, Ehrenkranz RA, Carlo WA, Shankaran S, Poole K, Wright LL 2003 Association between peak serum bilirubin and neurodevelopmental outcomes in extremely low birth weight infants. Pediatrics 112: 773–779
Van Overmeire B, Smets K, Lecoutere D, Van de Broek H, Weyler J, Degroote K, Langhendries JP 2000 A comparison of ibuprofen and indomethacin for closure of patent ductus arteriosus. N Engl J Med 343: 674–681
Ahlfors CE 2004 Effect of ibuprofen on bilirubin-albumin binding. J Pediatr 144: 386–388
Robinson PJ, Rapoport SI 1987 Binding effect of albumin on uptake of bilirubin by brain. Pediatrics 79: 553–558
Robinson PJ, Rapoport SI 1986 Kinetics of protein binding determine rates of uptake of drugs by brain. Am J Physiol 251: R1212–R1220
Ahlfors CE, Parker AE 2005 Evaluation of a model for brain bilirubin uptake in jaundiced newborns. Pediatr Res 58: 1175–1179
Ahlfors CE, Parker AE 2008 Unbound bilirubin concentration is associated with abnormal automated auditory brainstem response for jaundiced newborns. Pediatrics 121: 976–978
Cooper-Peel C, Brodersen R, Robertson A 1996 Does ibuprofen affect bilirubin-albumin binding in newborn infant serum?. Pharmacol Toxicol 79: 297–299
Silva RF, Rodrigues CM, Brites D 2002 Rat cultured neuronal and glial cells respond differently to toxicity of unconjugated bilirubin. Pediatr Res 51: 535–541
Hanko E, Hansen TW, Almaas R, Lindstad J, Rootwelt T 2005 Bilirubin induces apoptosis and necrosis in human NT2-N neurons. Pediatr Res 57: 179–184
Calligaris SD, Bellarosa C, Giraudi P, Wennberg RP, Ostrow JD, Tiribelli C 2007 Cytotoxicity is predicted by unbound and not total bilirubin concentration. Pediatr Res 62: 576–580
Brewer GJ 1995 Serum-free B27/neurobasal medium supports differentiated growth of neurons from the striatum, substantia nigra, septum, cerebral cortex, cerebellum, and dentate gyrus. J Neurosci Res 42: 674–683
Brodersen R, Stern L 1990 Deposition of bilirubin acid in the central nervous system–a hypothesis for the development of kernicterus. Acta Paediatr Scand 79: 12–19
Zucker SD, Goessling W 2000 Mechanism of hepatocellular uptake of albumin-bound bilirubin. Biochim Biophys Acta 1463: 197–208
Koh JY, Wie MB, Gwag BJ, Sensi SL, Canzoniero LM, Demaro J, Csernansky C, Choi DW 1995 Staurosporine-induced neuronal apoptosis. Exp Neurol 135: 153–159
Buhrer C, Atzpodien J, Oz S, Henze G 1991 Fosfomycin does not reduce cytostatic activity of cis-platinum against human osteosarcoma cell lines in vitro. Pediatr Hematol Oncol 8: 243–249
McDonald JW, Shapiro SM, Silverstein FS, Johnston MV 1998 Role of glutamate receptor-mediated excitotoxicity in bilirubin-induced brain injury in the Gunn rat model. Exp Neurol 150: 21–29
Hoffman DJ, Zanelli SA, Kubin J, Mishra OP, Delivoria-Papadopoulos M 1996 The in vivo effect of bilirubin on the N-methyl-d-aspartate receptor/ion channel complex in the brains of newborn piglets. Pediatr Res 40: 804–808
Hanko E, Hansen TW, Almaas R, Paulsen R, Rootwelt T 2006 Synergistic protection of a general caspase inhibitor and MK-801 in bilirubin-induced cell death in human NT2-N neurons. Pediatr Res 59: 72–77
Grojean S, Koziel V, Vert P, Daval JL 2000 Bilirubin induces apoptosis via activation of NMDA receptors in developing rat brain neurons. Exp Neurol 166: 334–341
Ostrow JD, Pascolo L, Tiribelli C 2003 Reassessment of the unbound concentrations of unconjugated bilirubin in relation to neurotoxicity in vitro. Pediatr Res 54: 98–104
Daood MJ, Watchko JF 2006 Calculated in vivo free bilirubin levels in the central nervous system of Gunn rat pups. Pediatr Res 60: 44–49
Aranda JV, Varvarigou A, Beharry K, Bansal R, Bardin C, Modanlou H, Papageorgiou A, Chemtob S 1997 Pharmacokinetics and protein binding of intravenous ibuprofen in the premature newborn infant. Acta Paediatr 86: 289–293
Van Overmeire B, Touw D, Schepens PJ, Kearns GL, van den Anker JN 2001 Ibuprofen pharmacokinetics in preterm infants with patent ductus arteriosus. Clin Pharmacol Ther 70: 336–343
Desfrere L, Zohar S, Morville P, Brunhes A, Chevret S, Pons G, Moriette G, Rey E, Treluyer JM 2005 Dose-finding study of ibuprofen in patent ductus arteriosus using the continual reassessment method. J Clin Pharm Ther 30: 121–132
Elsisi NS, Darling-Reed S, Lee EY, Oriaku ET, Soliman KF 2005 Ibuprofen and apigenin induce apoptosis and cell cycle arrest in activated microglia. Neurosci Lett 375: 91–96
Park EM, Cho BP, Volpe BT, Cruz MO, Joh TH, Cho S 2005 Ibuprofen protects ischemia-induced neuronal injury via up-regulating interleukin-1 receptor antagonist expression. Neuroscience 132: 625–631
Pender MP, Rist MJ 2001 Apoptosis of inflammatory cells in immune control of the nervous system: role of glia. Glia 36: 137–144
Iwata R, Kitagawa K, Zhang NY, Wu B, Inagaki C 2004 Non-steroidal anti-inflammatory drugs protect amyloid beta protein-induced increase in the intracellular Cl- concentration in cultured rat hippocampal neurons. Neurosci Lett 367: 156–159
Gasparini L, Ongini E, Wenk G 2004 Non-steroidal anti-inflammatory drugs (NSAIDs) in Alzheimer's disease: old and new mechanisms of action. J Neurochem 91: 521–536
Menzel JE, Kolarz G 1997 Modulation of nitric oxide synthase activity by ibuprofen. Inflammation 21: 451–461
Fotakis G, Timbrell JA 2006 In vitro cytotoxicity assays: comparison of LDH, neutral red, MTT and protein assay in hepatoma cell lines following exposure to cadmium chloride. Toxicol Lett 160: 171–177
Kosaka T, Fukaya K, Tsuboi S, Pu H, Ohno T, Tsuji T, Namba M 1996 Comparison of various methods of assaying the cytotoxic effects of ethanol on human hepatoblastoma cells (HUH-6 line). Acta Med Okayama 50: 151–156
Dore S, Snyder SH 1999 Neuroprotective action of bilirubin against oxidative stress in primary hippocampal cultures. Ann N Y Acad Sci 890: 167–172
Rodrigues CM, Sola S, Silva RF, Brites D 2002 Aging confers different sensitivity to the neurotoxic properties of unconjugated bilirubin. Pediatr Res 51: 112–118
Silva RF, Rodrigues CM, Brites D 2001 Bilirubin-induced apoptosis in cultured rat neural cells is aggravated by chenodeoxycholic acid but prevented by ursodeoxycholic acid. J Hepatol 34: 402–408
Acknowledgements
We thank Evelyn Strauss for technical assistance and help with the primary neuronal cultures, and Anne Gale for language editing.
Author information
Authors and Affiliations
Corresponding author
Additional information
This work was supported by internal university research grants (Charité, Universitätsmedizin Berlin, Germany).
Rights and permissions
About this article
Cite this article
Berns, M., Toennessen, M., Koehne, P. et al. Ibuprofen Augments Bilirubin Toxicity in Rat Cortical Neuronal Culture. Pediatr Res 65, 392–396 (2009). https://doi.org/10.1203/PDR.0b013e3181991511
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/PDR.0b013e3181991511
This article is cited by
-
Trichopus zeylanicus ameliorates ibuprofen inebriated hepatotoxicity and enteropathy: an insight into its modulatory impact on pro/anti-inflammatory cytokines and apoptotic signaling pathways
Inflammopharmacology (2022)
-
Ibuprofen Induces Mitochondrial-Mediated Apoptosis Through Proteasomal Dysfunction
Molecular Neurobiology (2016)
-
Patent ductus arteriosus in the preterm infant: to treat or not to treat?
Journal of Perinatology (2010)
