
Abstract
Reactive oxygen species (ROS) serve as cell signaling molecules for normal biologic processes. However, the generation of ROS can also provoke damage to multiple cellular organelles and processes, which can ultimately disrupt normal physiology. An imbalance between the production of ROS and the antioxidant defenses that protect cells has been implicated in the pathogenesis of a variety of diseases, such as cancer, asthma, pulmonary hypertension, and retinopathy. The nature of the injury will ultimately depend on specific molecular interactions, cellular locations, and timing of the insult. This review will outline the origins of endogenous and exogenously generated ROS. The molecular, cellular, pathologic, and physiologic targets will then be discussed with a particular emphasis on aspects relevant to child development. Finally, antioxidant defenses that scavenge ROS and mitigate associated toxicities will be presented, with a discussion of potential therapeutic approaches for the prevention and/or treatment of human diseases using enzymatic and nonenzymatic antioxidants.
Similar content being viewed by others

Main
Increasing evidence links early exposure to oxidative stress with potentially lifelong consequences (1). However, the role of reactive oxygen species (ROS) in biologic systems is entirely dependent on context: location, neighbors, and timing (2). ROS are oxygen ions [singlet oxygen, superoxide (O2·−)] or oxygen-containing radicals [hydroxyl, OH·−]. ROS and their reaction products [e.g. hydrogen peroxide (H2O2)] are increasingly recognized as signaling intermediates in their own right that can contribute to adaptive or maladaptive molecular responses (3). This review will focus on—1) origins of ROS: environment, cells, and cellular components; 2) molecular targets: classic and novel macromolecular targets and associated toxicity in infants and children; 3) antioxidant defenses: developmental regulation and vulnerabilities; and 4) antioxidant therapies: enzymatic and nonenzymatic approaches.
Origins of ROS
Oxygen has a unique molecular structure and is abundant within cells. It readily accepts free electrons generated by normal oxidative metabolism within the cell, producing ROS, such as O2·− and hydroxyl radical (HO·), as well as the oxidant H2O2. Processes causing uncoupling of electron transport can enhance the production of ROS, with mitochondria being a major source (4). However, other cellular components, such as endoplasmic reticulum-bound enzymes, cytoplasmic enzyme systems, and the surface of the plasma membrane, also contribute (5,6). Activity of multiple enzyme systems, such as the cytochrome P450 monoxygenase system, xanthine oxidoreductase, nitric oxide synthases, and several others involved in the inflammatory process (cyclooxygenase and lipoxygenase), can also increase the generation of ROS. Cellular production of O2·− and H2O2 can facilitate the formation of the more toxic and reactive HO· in the presence of reduced transition metals such as iron. Importantly, O2·− reacts rapidly with nitric oxide to form peroxynitrite (ONOO−), a strong nitrating and oxidizing compound (7). Such highly reactive species, such as HO· or ONOO−, can react with membrane lipids to cause more complex radicals by initiating lipid peroxidation.
In addition to ROS generated as a “byproduct” of cellular respiration, endogenous production of O2·− also arises from NADPH oxidases (NOX 1–3; typically at low levels in smooth muscle and vascular endothelium), dual oxidases 1 and 2, and NOX 4 (epithelial cells) (8). ROS are also important in the regulation of nitric oxide bioavailability, dramatically influencing airway and vascular reactivity (2). The burden of ROS can be further amplified by the presence of “free” metals, such as iron, copper, and manganese, which can be released from metalloprotein complexes. Although free iron (unbound to ferritin or heme, for example) has been documented in the circulating plasma of preterm newborns, the detrimental effects of free iron or other metals have not been definitively established in newborns (9,10). In contrast, indirect evidence has linked the presence of free iron with increased protein carbonyl formation in patients treated with high concentrations of supplemental oxygen. Failure to adequately sequester or store iron could be a developmental liability in premature infants with relative deficiencies in iron carriers, such as transferrin (11).
Molecular Targets: How ROS Damage Cells and Organs
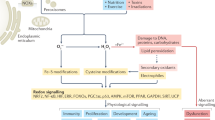
A delicate balance exists between ROS production and the antioxidant defenses that protect cells in vivo. This balance may become disturbed under conditions of hyperoxia, inflammation, or ischemia-reperfusion (excessive generation of ROS) or in the presence of limited or impaired antioxidant defenses. Multiple pathways involved in ROS-induced cell death have been proposed. ROS can cause direct injury to proteins, lipids, and nucleic acids, leading to cell death. Some of these pathways are illustrated in Fig. 1. For example, protein oxidation and nitrosylation (carbonyl, nitration, and nitrotyrosine formation) can impair a wide variety of enzymatic processes and growth factors that can result in marked cellular dysfunction (12). Lipid peroxidation has been linked to cell death through effects on cellular phospholipids (major cell membrane components) through activation of sphingomyelinase and release of ceramide, which activates apoptosis (13). Nucleic acid oxidation has been linked with physiologic and premature aging as well as DNA strand breaks, leading to necrosis and/or maladaptive apoptosis (14). The magnitude of these changes and the cell's ability to repair this damage determines whether the effects are adaptive or maladaptive.

ROS generation and detoxification in alveolar epithelium. Molecular oxygen first contacts the alveolus through the layer of surfactant phospholipids contained in the epithelial lining fluid, which is rich in glutathione and can, scavenge ROS. Under oxidative stress, ROS may be generated at the epithelial layer by DUOX and NOX4 that generate H2O2. SOD3 is poised to detoxify extracellular O2·−, although extracellular expression may be relatively deficient in newborns. Intracellularly, SOD2 in mitochondria detoxify O2·− generated during normal cellular respiration. The intracellular H2O2 burden is detoxified by peroxisome-bound catalase. Alveolar epithelium and other tissues may enhance generation of ROS in endothelial cells via NOX2,4, which can terminate NO-mediated reactions.
ROS at the proper locations and concentrations can also function as “2nd messengers” and activate multiple signal transduction pathways within the cell, facilitating the actions of growth factors, cytokines, and calcium signaling. ROS can activate c-Jun N-terminal kinase (possibly through production of lipid peroxide intermediates), a crucial mitogen-activated protein kinase, which then phosphorylates and releases two Bcl-2–related proteins that are normally sequestered within the cell (15,16). The release of these key proteins can directly activate Bax by causing dissociation from its cytoplasmic anchor. Bax is then free to translocate to the mitochondria, where it undergoes oligomerization and initiates the release of cytochrome c and other pro-death mediators into the cytosol.
Relatively high levels of O2·− are generated by NOX in phagocytes, such as neutrophils and macrophages (∼1000-fold higher than nonphagocytic cells), an essential process in bacterial killing. Blocking neutrophil influx in hyperoxia-exposed newborn rats mitigates oxidative DNA damage, HO· formation, and O2·− accumulation, while enhancing alveolar development (14,17–19). In experimental lung injury models, genetic ablation of NOX routinely reduces pulmonary ROS accumulation, but is not necessarily protective, because accompanying inflammation is worse in the NOX null mice (20,21). Loss of nonphagocytic NOX function may also impair physiologic signaling (22). The relevance of ROS generated by NOX depends on the organ system, with inhibition preventing radiation-induced oxidative stress in rat brain microvascular endothelium (23). In general, adult models of oxidative stress to the CNS show decreased injury with inhibition or genetic ablation of NOX activity (24). In contrast, newborn mice exposed to hypoxia-ischemia sustain worse injury with inhibition of NOX (e.g. NOX2 null mice), implying that endogenous O2·− signaling may have critical adaptive roles in different organ systems (25).
The brain and the lung have been most intensively studied as target organ systems prone to damage by ROS. In premature and full-term newborns, the control of cerebral perfusion is less tightly regulated, increasing vulnerability to reperfusion-type injury and oxidative stress. For example, microglial activation is believed to cause accumulation of markers of oxidation (e.g. nitrotyrosine and protein carbonyls) in oligodendrocytes, leading to the development of periventricular leukomalacia (26). Activation may also have secondary effects through neuronal excitotoxicity via effects on calcium flux. Most experimental studies have focused on ischemia-reperfusion models, with pretreatment with antioxidants or free radical scavengers typically reducing apoptosis (e.g. less DNA fragmentation and caspase expression) and ameliorating histologic evidence of brain injury (27,28). Recently, studies of hyperoxia in newborn rats have also implicated ROS in causing neuronal cell death. In vitro exposure to high-oxygen atmospheres induces apoptosis in oligodendroglial cells in a developmentally dependent pattern, which is prevented by inhibition of lipoxygenase, with decreased expression of myelin basic protein in vivo in hyperoxia-exposed rat pups (29).
The developing retina is particularly prone to ROS-mediated damage that contributes to retinopathy of prematurity in preterm infants (30). Vascular growth into the developing posterior retina is normally driven by redox-sensitive pathways that up-regulate VEGF. After birth, the marked increase in systemic oxygen tensions in the preterm newborn suppresses VEGF production. This occurs in conjunction with impairment in autoregulation of retinal blood flow as well as a relative deficiency of antioxidants in the immature retina (31). In the absence of VEGF (and other factors), angiogenic budding stops, and apoptosis of developing vessels occurs secondary to the formation of reactive oxygen and nitrogen species (32–34). Endogenous generation of ROS through NOX may be critical to this pathway, because its pharmacologic inhibition prevents retinopathy of prematurity in a newborn rat model (35). The second phase of retinopathy of prematurity occurs after birth, when the avascular retina continues to grow, overreaching its blood supply. This results in local tissue hypoxia, increased VEGF release, and an abnormal neovascular response. Once again, this process involves the formation of ROS and may be amenable to reductions in exposure to oxygen as well as treatment with antioxidants (36,37).
Similar mechanisms may be at play in ROS-induced damage to the immature postnatal, developing pulmonary system, where both epithelial and endothelial cells may be damaged. Pulmonary epithelial DNA oxidation (14), accumulation of HO· (19), lipid peroxidation (38), and protein oxidation (39) in whole lung have all been demonstrated in experimental models of bronchopulmonary dysplasia (BPD). In human BPD, studies do strongly support a role for ROS-mediated damage. Plasma 3-nitrotyrosine, a footprint of ONOO− formation, and protein carbonyls, a marker of protein oxidation, are elevated in premature newborns at highest risk of developing BPD (40). ROS may inactivate antioxidant enzymes, with oxidized or nitrated proteins critical to lung function having been identified (41). The weight of the evidence implicates ROS in the development of impaired lung development in BPD (9,42).
Because exposure to more moderate oxygen concentrations are associated with modern-day BPD, the disruption to mesenchymal-epithelial-endothelial signaling, rather than acute cell necrosis or apoptosis induced by ROS, may be more critical. The “new” BPD is characterized by milder exposure to oxidative stress and mechanical injury, but at an earlier stage of pulmonary development, which ultimately causes alveolar hypoplasia (43). The inactivation of NO signaling, which is required for normal alveolar development, is one likely candidate pathway, either through direct inactivation (ONOO− formation) or through indirect effects on endogenous NO production (44,45). In some (46,47), but not all, (48,49) experimental animal models of BPD, treatment with inhaled NO significantly protects alveolar development. Clinical trials of inhaled NO to prevent BPD have also had mixed results (50–53), possibly due to differences in patient selection and treatment strategies as well as inactivation of NO by ROS. Because cGMP is a major target of NO action, type V phosphodiesterase inhibition would be expected to also enhance alveolar development, which indeed has been shown in a newborn rat model of BPD (54). Further support for this concept is suggested by strategies aimed at interfering with O2−–mediated inactivation of NO using exogenous recombinant human CuZnSOD (rhSOD). In newborn lambs with pulmonary hypertension, treatment with inhaled NO in conjunction with rhSOD results in enhanced NO signaling, significant improvements in oxygenation, and marked reductions in oxidation in the lung (55). Clinical trials of inhaled NO in conjunction with rhSOD administration for the treatment of pulmonary hypertension in newborn infants are currently being planned.
Antioxidant Defenses: Developmental Regulation and Vulnerabilities
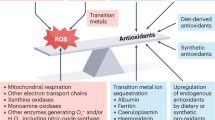
The vulnerability of target molecules, cellular compartments, and organ systems to ROS-mediated pathways depends on the local redox milieu, which, in turn, depends on developmental regulation of antioxidants. Several foci of ROS generation are spatially adjacent to opposing antioxidant enzymatic systems within subcellular compartments, as illustrated in Fig. 2. Tight temporo-spatial control of antioxidant expression has been linked to normal control of apoptosis in physiologic development. For example, opposition to oxidative signaling by use of antioxidant mimetics has been shown to prevent the physiologic apoptosis required for normal vertebrate limb development (56).

ROS, macromolecular damage, altered signaling. ROS damage DNA through strand breaks and base oxidation that, if unrepaired, induces apoptosis or oncosis. Protein oxidation and nitration damage antioxidant enzymes, surfactant proteins, and anti-inflammatory pathways that can further propagate maladaptive inflammation. Lipid peroxidation products generate pro-inflammatory prostanoids, and can generate further radical formation through lipid chain reactions, possibly releasing damaging enzymes packaged in cellular organelles. Direct effects of ROS on signaling pathways include redox-sensitive transcription factors—e.g. HIF, Nrf-2, and NF-κB—as well as indirect effects through inactivation of NO-based signaling.
Gestational dependence of enzymatic antioxidants has been recognized for decades, as has the developmental regulation of oxygen-dependent signaling (previously reviewed in this series by Maltepe and Saugstad) (57,58). The cellular, subcellular, and tissue-specific expression of antioxidant enzymes, such as superoxide dismutases (SOD), catalase, glutathione peroxidases, and peroxiredoxin largely determine the relative vulnerabilities of tissues and cells to ROS-mediated injury (59,60). However, oxidative stress may actually regulate antioxidant capacity, with newborn rats demonstrating up-regulation of glutathione peroxidase catalase, and CuZn (cytosolic) SOD expression and activity in response to hyperoxia (61).
Because enzymatic antioxidants are gestationally regulated, premature newborns would be expected to have decreased expression relative to full-term newborns, and this has been demonstrated in most animal models (58,59,62,63). Susceptibility of the premature infant to ROS-mediated damage also depends on the expression and activity of many of these antioxidant enzymes, with heme oxygenase-1 and thioredoxin mRNA expression levels increased immediately after birth in both preterm and term newborns (64,65). Hypoxia may generate ROS that directly or indirectly stimulate hypoxia-inducible factors important in lung or brain development (66,67). The contribution of these induced responses may vary depending on the nature and magnitude of the oxidative stress. Whether this gestation-dependent expression/induction of antioxidants is adequate and actually involved in human diseases is not completely clear. Some antioxidant levels—glutathione, ascorbate, and urate—have been analyzed in tracheal aspirates and found to be poorly predictive of the risk of developing BPD (68). However, other antioxidants (e.g. SOD) may be more important in view of what has been found in animal model systems as well as clinical specimens obtained from newborn infants (69).
Although nonenzymatic antioxidants are also depleted in conditions characterized by ROS-mediated stress, the interpretation of these measurements is quite complex (70). For example, “low” vitamin E levels in premature newborns were interpreted as representing a deficient state; whether or not this is the case actually depends on the specific target cell or organ and on the actual ROS milieu (71). In contrast, glutathione may be deficient in premature infants because of excessive oxidization by ROS coupled with reduced glutathione reductase reaction with the electron acceptor NADPH (72). Finally, melatonin acts as an antioxidant in the retina and brain (73), and its cyclic production is disrupted in premature infants (74), possibly increasing the risk of ROS-mediated damage.
Antioxidant Therapies: Enzymatic and Nonenzymatic Approaches
Although the use of supplemental antioxidants represents a logical strategy to prevent or ameliorate lung injury from excess generation of ROS, caution must be exercised because ROS are critically important second messengers in various cell signaling pathways that control normal cellular functions. In addition, intracellular generation of ROS is important in bacterial killing by alveolar macrophages and neutrophils, and antioxidants may interfere with this process and contribute to worsening tissue injury.
Multiple cell culture models have suggested that overexpression of antioxidants prevents ROS-induced injury. Ilizarov et al. (75) generated stable cell lines overexpressing MnSOD and/or catalase (1.5- to 2-fold increase in activity) and then exposed them to 95% O2 for 10 d. Significantly, more cells overexpressing MnSOD were viable (∼40%) compared with cells overexpressing catalase alone or control cells (∼10%). Overexpression of catalase with MnSOD had a small additional benefit, suggesting that scavenging O2·− is the important rate-limiting step. Overexpression of either MnSOD or CuZnSOD also reversed the growth inhibitory effects of hyperoxia, with optimal protection from hyperoxic injury occurring with 1.5- to 3-fold increases in activity (76). Prevention of mitochondrial oxidation seemed to be a critical factor, because markers of mitochondrial function and cell survival correlated directly with the extent of mitochondrial localization of antioxidant activity and not overall activity within the cell (77). Overexpression of SOD not only reduced ROS production, but also mitigated the activation of the JNK/AP-1 pathway (78). Activation of this MAPK signal transduction pathway has been implicated in the pathogenesis of ROS-induced mitochondrial injury and apoptotic cell death. Finally, bacterial infection and associated inflammation have been shown to significantly increase ROS production. Exposure of lung epithelial cells (both airway and alveolar), monocytes, and macrophages to hyperoxia for as little as 24 h is associated with significant increases in bacterial adherence and IL-8 production as well as impaired phagocytosis and bacterial clearance, with overexpression of SOD having significant beneficial effects (79–81). Because nosocomial infection is a predictor of BPD (82), antioxidant therapy could also be protective through this mechanism.
Other data demonstrating the efficacy of SOD in preventing hyperoxia-induced lung injury come from studies of genetically engineered mice. Transgenic mice lacking MnSOD die within the first 10 d of life in room air, whereas mice lacking CuZn or EC-SOD have reduced survival and more lung injury in response to ROS, but a normal lifespan (83–85). In contrast, transgenic mice overexpressing MnSOD in alveolar type II cells are able to survive longer with significantly less lung injury in hyperoxia compared with wild-type controls (86). In addition, newborn EC-SOD transgenic (SP-C promoter driven) mice exposed to hyperoxia showed significantly less pulmonary neutrophil influx and reduced glutathione, with preservation of alveolar development compared with wild-type littermates (60). Transgenic mice had significantly less pulmonary neutrophil influx and oxidized glutathione at 7 d, preservation of alveolar surface and volume density, and preserved differentiation of type I alveolar epithelium, compared with wild-type littermates. Taken together, these data indicate that SOD is critically important in preventing hyperoxia-induced lung injury and preserving normal alveolar architecture.
The antioxidant vitamins, ascorbic acid (vitamin C) and α-tocopherol (vitamin E), are known to inhibit ROS-induced lipid peroxidation. Berger et al. (87) studied the administration of high-dose antioxidant vitamins in a premature baboon model of BPD compared with standard antioxidant vitamin supplementation. Although higher doses significantly raised vitamin C and E concentrations in plasma and the lung, no protective effects could be demonstrated. These studies question whether raising antioxidant vitamin concentrations alone will be effective in preventing ROS-induced injury in high-risk preterm infants.
Supplementation of vitamins A, C, and E has also been studied in preterm infants in an attempt to prevent with ROS-induced injury. Concentrations of vitamin A (i.e. retinol) may be deficient in very low birth weight infants, presumably from increased absorption of parenteral vitamin A into the i.v. tubing or from higher nutritional requirements (88). A multicenter trial of high-dose vitamin A supplementation in premature infants found a small (7%), but statistically significant reduction in the incidence of BPD (89). However, follow-up of treated infants did not demonstrate any long-term benefits of vitamin A in reducing chronic respiratory morbidity (90). Vitamin C has both oxidant and antioxidant activities and is thought to contribute to the regeneration of membrane-bound α-tocopherol (91). Although preterm infants may be relatively deficient in vitamin E, randomized controlled trials have consistently failed to demonstrate a significant benefit of α-tocopherol in preterm infants (92,93). Of concern, is that pharmacologic concentrations of vitamin E were associated with an increased risk of sepsis and necrotizing enterocolitis, precluding the routine use of these doses in high-risk preterm infants (94).
n-Acetylcysteine (NAC) is a source of the essential amino acid l-cysteine and a precursor of the antioxidant glutathione. A multicenter, double-blind trial of NAC was conducted in 391 ventilated, extremely low birth weight infants. Infants were randomized by 36 h of age to receive 16–32 mg/kg/d of NAC or placebo i.v. for 6 d (95). The study showed no reduction in survival or the incidence or severity of BPD at 36 wk corrected age or improved pulmonary function when the infants were studied at term (96).
A multicenter, randomized trial of prophylactic rhSOD has been performed to determine whether intratracheal treatment significantly reduced the incidence of BPD and improved pulmonary outcome at 1-y corrected age (97). Three hundred two intubated, premature infants (600–1200 g at birth) received either intratracheal rhSOD (5 mg/kg) or placebo every 48 h (as long as intubation was required) up to 1 mo of age. Although there were no differences in the incidence of death or BPD, 37% of placebo-treated infants had repeated episodes of wheezing or other respiratory illness severe enough to require treatment with asthma medications (e.g. bronchodilators and corticosteroids) compared with 24% of rhSOD treated infants at 1-y corrected age. In the highest risk infants, <27 wk gestation, 42% treated with placebo received asthma medications compared with 19% of rhSOD-treated infants. rhSOD was also associated with a 55% decrease in emergency department visits and a 44% decrease in subsequent hospitalizations. This study demonstrates that rhSOD may reduce ROS-induced pulmonary injury, although this may not be readily apparent when only evaluating early outcomes based on current BPD definitions. Future studies using long-term outcome variables may be needed to more definitively determine whether treatments to scavenge ROS are effective.
Summary
Oxidative stress, particularly, in the preterm newborn, arises in multiple organ systems and subcellular compartments. This occurs due to inadequate detoxifying mechanisms such as inducible antioxidant enzymes, glutathione stores, and nutritional antioxidants. Oxidative molecular damage to DNA can arrest appropriately timed proliferation and differentiation and damage to lipids in cell membranes, and key regulatory enzymes can provoke maladaptive inflammatory responses that can amplify the initial injuries. More subtle effects on ROS-mediated signaling and depletion of NO available for endogenous proangiogenic signaling can further contribute to disrupted organ development, including excitotoxic neuronal damage. Although these aspects have suggested the rationale for antioxidant therapy, its uses in the prevention of BPD, ROP, or brain injury in preterm newborns has not yet yielded unequivocal success. Further studies aimed at superior targeting to improve the therapeutic index of antioxidants will be necessary.
Abbreviations
- BPD:
-
bronchopulmonary dysplasia
- H2O2:
-
hydrogen peroxide
- HO·:
-
hydroxyl radical
- NAC:
-
n-acetylcysteine
- NOX:
-
NAD(P)H oxidase
- ROS:
-
reactive oxygen species
- SOD:
-
superoxide dismutase
- O2·−:
-
superoxide
References
Sola A, Rogido MR, Deulofeut R 2007 Oxygen as a neonatal health hazard: call for detente in clinical practice. Acta Paediatr 96: 801–812
Lambeth JD 2007 Nox enzymes, ROS, and chronic disease: an example of antagonistic pleiotropy. Free Radic Biol Med 43: 332–347
Covarrubias L, Hernandez-Garcia D, Schnabel D, Salas-Vidal E, Castro-Obregon S 2008 Function of reactive oxygen species during animal development: passive or active?. Dev Biol 320: 1–11
Gruber J, Schaffer S, Halliwell B 2008 The mitochondrial free radical theory of ageing—where do we stand?. Front Biosci 13: 6554–6579
Davydov DR 2001 Microsomal monooxygenase in apoptosis: another target for cytochrome c signaling?. Trends Biochem Sci 26: 155–160
Sumimoto H 2008 Structure, regulation and evolution of Nox-family NADPH oxidases that produce reactive oxygen species. FEBS J 275: 3249–3277
Beckman JS, Beckman TW, Chen J, Marshall PA, Freeman BA 1990 Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc Natl Acad Sci USA 87: 1620–1624
van der Vliet A 2008 NADPH oxidases in lung biology and pathology: host defense enzymes, and more. Free Radic Biol Med 44: 938–955
Saugstad OD 2003 Bronchopulmonary dysplasia-oxidative stress and antioxidants. Semin Neonatol 8: 39–49
Evans PJ, Evans R, Kovar IZ, Holton AF, Halliwell B 1992 Bleomycin-detectable iron in the plasma of premature and full-term neonates. FEBS Lett 303: 210–212
Lindeman JH, Lentjes EG, van Zoeren-Grobben D, Berger HM 2000 Postnatal changes in plasma ceruloplasmin and transferrin antioxidant activities in preterm babies. Biol Neonate 78: 73–76
Stadtman ER, Levine RL 2000 Protein oxidation. Ann N Y Acad Sci 899: 191–208
Fruhwirth GO, Hermetter A 2008 Mediation of apoptosis by oxidized phospholipids. Subcell Biochem 49: 351–367
Auten RL, Whorton MH, Nicholas Mason S 2002 Blocking neutrophil influx reduces DNA damage in hyperoxia-exposed newborn rat lung. Am J Respir Cell Mol Biol 26: 391–397
Parinandi NL, Kleinberg MA, Usatyuk PV, Cummings RJ, Pennathur A, Cardounel AJ, Zweier JL, Garcia JG, Natarajan V 2003 Hyperoxia-induced NAD(P)H oxidase activation and regulation by MAP kinases in human lung endothelial cells. Am J Physiol Lung Cell Mol Physiol 284: L26–L38
Lei K, Davis RJ 2003 JNK phosphorylation of Bim-related members of the Bcl2 family induces Bax-dependent apoptosis. Proc Natl Acad Sci USA 100: 2432–2437
Belik J, Jankov RP, Pan J, Yi M, Chaudhry I, Tanswell AK 2004 Chronic O2 exposure in the newborn rat results in decreased pulmonary arterial nitric oxide release and altered smooth muscle response to isoprostane. J Appl Physiol 96: 725–730
Auten RL Jr, Mason SN, Tanaka DT, Welty-Wolf K, Whorton MH 2001 Anti-neutrophil chemokine preserves alveolar development in hyperoxia-exposed newborn rats. Am J Physiol Lung Cell Mol Physiol 281: L336–L344
Liao L, Ning Q, Li Y, Wang W, Wang A, Wei W, Liu X, Auten RL, Tanswell AK, Luo X 2006 CXCR2 blockade reduces radical formation in hyperoxia-exposed newborn rat lung. Pediatr Res 60: 299–303
Gao XP, Standiford TJ, Rahman A, Newstead M, Holland SM, Dinauer MC, Liu QH, Malik AB 2002 Role of NADPH oxidase in the mechanism of lung neutrophil sequestration and microvessel injury induced by Gram-negative sepsis: studies in p47phox−/− and gp91phox−/− mice. J Immunol 168: 3974–3982
Yao H, Edirisinghe I, Yang SR, Rajendrasozhan S, Kode A, Caito S, Adenuga D, Rahman I 2008 Genetic ablation of NADPH oxidase enhances susceptibility to cigarette smoke-induced lung inflammation and emphysema in mice. Am J Pathol 172: 1222–1237
Chitano P, Wang L, Mason SN, Auten RL, Potts EN, Foster WM, Sturrock A, Kennedy TP, Hoidal JR, Murphy TM 2008 Airway smooth muscle relaxation is impaired in mice lacking the p47phox subunit of NAD(P)H oxidase. Am J Physiol Lung Cell Mol Physiol 294: L139–L148
Collins-Underwood JR, Zhao W, Sharpe JG, Robbins ME 2008 NADPH oxidase mediates radiation-induced oxidative stress in rat brain microvascular endothelial cells. Free Radic Biol Med 45: 929–938
Infanger DW, Sharma RV, Davisson RL 2006 NADPH oxidases of the brain: distribution, regulation, and function. Antioxid Redox Signal 8: 1583–1596
Doverhag C, Keller M, Karlsson A, Hedtjarn M, Nilsson U, Kapeller E, Sarkozy G, Klimaschewski L, Humpel C, Hagberg H, Simbruner G, Gressens P, Savman K 2008 Pharmacological and genetic inhibition of NADPH oxidase does not reduce brain damage in different models of perinatal brain injury in newborn mice. Neurobiol Dis 31: 133–144
Khwaja O, Volpe JJ 2008 Pathogenesis of cerebral white matter injury of prematurity. Arch Dis Child Fetal Neonatal Ed 93: F153–F161
Miura S, Ishida-Nakajima W, Ishida A, Kawamura M, Ohmura A, Oguma R, Sato Y, Takahashi T 2009 Ascorbic acid protects the newborn rat brain from hypoxic-ischemia. Brain Dev 31: 307–317
Yasuoka N, Nakajima W, Ishida A, Takada G 2004 Neuroprotection of edaravone on hypoxic-ischemic brain injury in neonatal rats. Brain Res Dev Brain Res 151: 129–139
Gerstner B, DeSilva TM, Genz K, Armstrong A, Brehmer F, Neve RL, Felderhoff-Mueser U, Volpe JJ, Rosenberg PA 2008 Hyperoxia causes maturation-dependent cell death in the developing white matter. J Neurosci 28: 1236–1245
Pierce EA, Foley ED, Smith LE 1996 Regulation of vascular endothelial growth factor by oxygen in a model of retinopathy of prematurity. Arch Ophthalmol 114: 1219–1228
Hardy P, Dumont I, Bhattacharya M, Hou X, Lachapelle P, Varma DR, Chemtob S 2000 Oxidants, nitric oxide and prostanoids in the developing ocular vasculature: a basis for ischemic retinopathy. Cardiovasc Res 47: 489–509
Gu X, Samuel S, El-Shabrawey M, Caldwell RB, Bartoli M, Marcus DM, Brooks SE 2002 Effects of sustained hyperoxia on revascularization in experimental retinopathy of prematurity. Invest Ophthalmol Vis Sci 43: 496–502
Gu X, El-Remessy AB, Brooks SE, Al-Shabrawey M, Tsai NT, Caldwell RB 2003 Hyperoxia induces retinal vascular endothelial cell apoptosis through formation of peroxynitrite. Am J Physiol Cell Physiol 285: C546–C554
Saugstad OD 2006 Oxygen and retinopathy of prematurity. J Perinatol 26: S46–S50; discussion S63–S44
Saito Y, Geisen P, Uppal A, Hartnett ME 2007 Inhibition of NAD(P)H oxidase reduces apoptosis and avascular retina in an animal model of retinopathy of prematurity. Mol Vis 13: 840–853
Raju TN, Langenberg P, Bhutani V, Quinn GE 1997 Vitamin E prophylaxis to reduce retinopathy of prematurity: a reappraisal of published trials. J Pediatr 131: 844–850
Chow LC, Wright KW, Sola A 2003 Can changes in clinical practice decrease the incidence of severe retinopathy of prematurity in very low birth weight infants?. Pediatrics 111: 339–345
Luo X, Sedlackova L, Belcastro R, Cabacungan J, Lye SJ, Tanswell AK 1999 Effect of the 21-aminosteroid U74389G on oxygen-induced free radical production, lipid peroxidation, and inhibition of lung growth in neonatal rats. Pediatr Res 46: 215–223
Vozzelli MA, Mason SN, Whorton MH, Auten RL Jr 2004 Antimacrophage chemokine treatment prevents neutrophil and macrophage influx in hyperoxia-exposed newborn rat lung. Am J Physiol Lung Cell Mol Physiol 286: L488–L493
Ballard PL, Truog WE, Merrill JD, Gow A, Posencheg M, Golombek SG, Parton LA, Luan X, Cnaan A, Ballard RA 2008 Plasma biomarkers of oxidative stress: relationship to lung disease and inhaled nitric oxide therapy in premature infants. Pediatrics 121: 555–561
Zhu S, Haddad IY, Matalon S 1996 Nitration of surfactant protein A (SP-A) tyrosine residues results in decreased mannose binding ability. Arch Biochem Biophys 333: 282–290
Martin RJ, Walsh MC 2004 Pre- and postnatal factors in chronic lung disease: implications for management. Paediatr Respir Rev 5: S235–S240
Jobe AJ 1999 The new BPD: an arrest of lung development. Pediatr Res 46: 641–643
Han RN, Stewart DJ 2006 Defective lung vascular development in endothelial nitric oxide synthase-deficient mice. Trends Cardiovasc Med 16: 29–34
Afshar S, Gibson LL, Yuhanna IS, Sherman TS, Kerecman JD, Grubb PH, Yoder BA, McCurnin DC, Shaul PW 2003 Pulmonary NO synthase expression is attenuated in a fetal baboon model of chronic lung disease. Am J Physiol Lung Cell Mol Physiol 284: L749–L758
Tang JR, Markham NE, Lin YJ, McMurtry IF, Maxey A, Kinsella JP, Abman SH 2004 Inhaled nitric oxide attenuates pulmonary hypertension and improves lung growth in infant rats after neonatal treatment with a VEGF receptor inhibitor. Am J Physiol Lung Cell Mol Physiol 287: L344–L351
ter Horst SA, Walther FJ, Poorthuis BJ, Hiemstra PS, Wagenaar GT 2007 Inhaled nitric oxide attenuates pulmonary inflammation and fibrin deposition and prolongs survival in neonatal hyperoxic lung injury. Am J Physiol Lung Cell Mol Physiol 293: L35–L44
McCurnin DC, Pierce RA, Chang LY, Gibson LL, Osborne-Lawrence S, Yoder BA, Kerecman JD, Albertine KH, Winter VT, Coalson JJ, Crapo JD, Grubb PH, Shaul PW 2005 Inhaled NO improves early pulmonary function and modifies lung growth and elastin deposition in a baboon model of neonatal chronic lung disease. Am J Physiol Lung Cell Mol Physiol 288: L450–L459
Auten RL, Mason SN, Whorton MH, Lampe WR, Foster WM, Goldberg RN, Li B, Stamler JS, Auten KM 2007 Inhaled ethyl nitrite prevents hyperoxia-impaired postnatal alveolar development in newborn rats. Am J Respir Crit Care Med 176: 291–299
Schreiber MD, Gin-Mestan K, Marks JD, Huo D, Lee G, Srisuparp P 2003 Inhaled nitric oxide in premature infants with the respiratory distress syndrome. N Engl J Med 349: 2099–2107
Van Meurs KP, Wright LL, Ehrenkranz RA, Lemons JA, Ball MB, Poole WK, Perritt R, Higgins RD, Oh W, Hudak ML, Laptook AR, Shankaran S, Finer NN, Carlo WA, Kennedy KA, Fridriksson JH, Steinhorn RH, Sokol GM, Konduri GG, Aschner JL, Stoll BJ, D'Angio CT, Stevenson DK 2005 Inhaled nitric oxide for premature infants with severe respiratory failure. N Engl J Med 353: 13–22
Kinsella JP, Cutter GR, Walsh WF, Gerstmann DR, Bose CL, Hart C, Sekar KC, Auten RL, Bhutani VK, Gerdes JS, George TN, Southgate WM, Carriedo H, Couser RJ, Mammel MC, Hall DC, Pappagallo M, Sardesai S, Strain JD, Baier M, Abman SH 2006 Early inhaled nitric oxide therapy in premature newborns with respiratory failure. N Engl J Med 355: 354–364
Ballard RA, Truog WE, Cnaan A, Martin RJ, Ballard PL, Merrill JD, Walsh MC, Durand DJ, Mayock DE, Eichenwald EC, Null DR, Hudak ML, Puri AR, Golombek SG, Courtney SE, Stewart DL, Welty SE, Phibbs RH, Hibbs AM, Luan X, Wadlinger SR, Asselin JM, Coburn CE 2006 Inhaled nitric oxide in preterm infants undergoing mechanical ventilation. N Engl J Med 355: 343–353
Ladha F, Bonnet S, Eaton F, Hashimoto K, Korbutt G, Thebaud B 2005 Sildenafil improves alveolar growth and pulmonary hypertension in hyperoxia-induced lung injury. Am J Respir Crit Care Med 172: 750–756
Lakshminrusimha S, Russell JA, Wedgwood S, Gugino SF, Kazzaz JA, Davis JM, Steinhorn RH 2006 Superoxide dismutase improves oxygenation and reduces oxidation in neonatal pulmonary hypertension. Am J Respir Crit Care Med 174: 1370–1377
Schnabel D, Salas-Vidal E, Narvaez V, Sanchez-Carbente Mdel R, Hernandez-Garcia D, Cuervo R, Covarrubias L 2006 Expression and regulation of antioxidant enzymes in the developing limb support a function of ROS in interdigital cell death. Dev Biol 291: 291–299
Frank L, Groseclose EE 1984 Preparation for birth into an O2-rich environment: the antioxidant enzymes in the developing rabbit lung. Pediatr Res 18: 240–244
Asikainen TM, Raivio KO, Saksela M, Kinnula VL 1998 Expression and developmental profile of antioxidant enzymes in human lung and liver. Am J Respir Cell Mol Biol 19: 942–949
Mamo LB, Suliman HB, Giles BL, Auten RL, Piantadosi CA, Nozik-Grayck E 2004 Discordant extracellular superoxide dismutase expression and activity in neonatal hyperoxic lung. Am J Respir Crit Care Med 170: 313–318
Ahmed MN, Suliman HB, Folz RJ, Nozik-Grayck E, Golson ML, Mason SN, Auten RL 2003 Extracellular superoxide dismutase protects lung development in hyperoxia-exposed newborn mice. Am J Respir Crit Care Med 167: 400–405
Clerch LB, Massaro D 1992 Rat lung antioxidant enzymes: differences in perinatal gene expression and regulation. Am J Physiol 263: L466–L470
Chen W, Hunt DM, Lu H, Hunt RC 1999 Expression of antioxidant protective proteins in the rat retina during prenatal and postnatal development. Invest Ophthalmol Vis Sci 40: 744–751
Clerch LB, Wright AE, Coalson JJ 1996 Lung manganese superoxide dismutase protein expression increases in the baboon model of bronchopulmonary dysplasia and is regulated at a posttranscriptional level. Pediatr Res 39: 253–258
Maroti Z, Katona M, Orvos H, Nemeth I, Farkas I, Turi S 2007 Heme oxygenase-1 expression in premature and mature neonates during the first week of life. Eur J Pediatr 166: 1033–1038
Das KC, Guo XL, White CW 1999 Induction of thioredoxin and thioredoxin reductase gene expression in lungs of newborn primates by oxygen. Am J Physiol 276: L530–L539
Asikainen TM, White CW 2005 Antioxidant defenses in the preterm lung: role for hypoxia-inducible factors in BPD?. Toxicol Appl Pharmacol 203: 177–188
Tomita S, Ueno M, Sakamoto M, Kitahama Y, Ueki M, Maekawa N, Sakamoto H, Gassmann M, Kageyama R, Ueda N, Gonzalez FJ, Takahama Y 2003 Defective brain development in mice lacking the Hif-1alpha gene in neural cells. Mol Cell Biol 23: 6739–6749
Collard KJ, Godeck S, Holley JE, Quinn MW 2004 Pulmonary antioxidant concentrations and oxidative damage in ventilated premature babies. Arch Dis Child Fetal Neonatal Ed 89: F412–F416
Jankov RP, Negus A, Tanswell AK 2001 Antioxidants as therapy in the newborn: some words of caution. Pediatr Res 50: 681–687
Schock BC, Sweet DG, Halliday HL, Young IS, Ennis M 2001 Oxidative stress in lavage fluid of preterm infants at risk of chronic lung disease. Am J Physiol Lung Cell Mol Physiol 281: L1386–L1391
Phelps DL 1988 The role of vitamin E therapy in high-risk neonates. Clin Perinatol 15: 955–963
Lavoie JC, Chessex P 1997 Gender and maturation affect glutathione status in human neonatal tissues. Free Radic Biol Med 23: 648–657
Siu AW, Maldonado M, Sanchez-Hidalgo M, Tan DX, Reiter RJ 2006 Protective effects of melatonin in experimental free radical-related ocular diseases. J Pineal Res 40: 101–109
Kennaway DJ, Stamp GE, Goble FC 1992 Development of melatonin production in infants and the impact of prematurity. J Clin Endocrinol Metab 75: 367–369
Ilizarov AM, Koo HC, Kazzaz JA, Mantell LL, Li Y, Bhapat R, Pollack S, Horowitz S, Davis JM 2001 Overexpression of manganese superoxide dismutase protects lung epithelial cells against oxidant injury. Am J Respir Cell Mol Biol 24: 436–441
Koo HC, Davis JM, Li Y, Hatzis D, Opsimos H, Pollack S, Strayer MS, Ballard PL, Kazzaz JA 2005 Effects of transgene expression of superoxide dismutase and glutathione peroxidase on pulmonary epithelial cell growth in hyperoxia. Am J Physiol Lung Cell Mol Physiol 288: L718–L726
Arita Y, Harkness SH, Kazzaz JA, Koo HC, Joseph A, Melendez JA, Davis JM, Chander A, Li Y 2006 Mitochondrial localization of catalase provides optimal protection from H2O2-induced cell death in lung epithelial cells. Am J Physiol Lung Cell Mol Physiol 290: L978–L986
Joseph A, Li Y, Koo HC, Davis JM, Pollack S, Kazzaz JA 2008 Superoxide dismutase attenuates hyperoxia-induced interleukin-8 induction via AP-1. Free Radic Biol Med 45: 1143–1149
Arita Y, Kazzaz JA, Joseph A, Koo HC, Li Y, Davis JM 2007 Antioxidants improve antibacterial function in hyperoxia-exposed macrophages. Free Radic Biol Med 42: 1517–1523
Arita Y, Joseph A, Koo HC, Li Y, Palaia TA, Davis JM, Kazzaz JA 2004 Superoxide dismutase moderates basal and induced bacterial adherence and interleukin-8 expression in airway epithelial cells. Am J Physiol Lung Cell Mol Physiol 287: L1199–L1206
Walti H, Nicolas-Robin A, Assous MV, Polla BS, Bachelet M, Davis JM 2002 Effects of exogenous surfactant and recombinant human copper-zinc superoxide dismutase on oxygen-dependent antimicrobial defenses. Biol Neonate 82: 96–102
Bancalari E 2001 Changes in the pathogenesis and prevention of chronic lung disease of prematurity. Am J Perinatol 18: 1–9
Melov S, Coskun P, Patel M, Tuinstra R, Cottrell B, Jun AS, Zastawny TH, Dizdaroglu M, Goodman SI, Huang TT, Miziorko H, Epstein CJ, Wallace DC 1999 Mitochondrial disease in superoxide dismutase 2 mutant mice. Proc Natl Acad Sci USA 96: 846–851
Carlsson LM, Jonsson J, Edlund T, Marklund SL 1995 Mice lacking extracellular superoxide dismutase are more sensitive to hyperoxia. Proc Natl Acad Sci USA 92: 6264–6268
Reaume AG, Elliott JL, Hoffman EK, Kowall NW, Ferrante RJ, Siwek DF, Wilcox HM, Flood DG, Beal MF, Brown RH Jr, Scott RW, Snider WD 1996 Motor neurons in Cu/Zn superoxide dismutase-deficient mice develop normally but exhibit enhanced cell death after axonal injury. Nat Genet 13: 43–47
Wispe JR, Warner BB, Clark JC, Dey CR, Neuman J, Glasser SW, Crapo JD, Chang LY, Whitsett JA 1992 Human Mn-superoxide dismutase in pulmonary epithelial cells of transgenic mice confers protection from oxygen injury. J Biol Chem 267: 23937–23941
Berger TM, Frei B, Rifai N, Avery ME, Suh J, Yoder BA, Coalson JJ 1998 Early high dose antioxidant vitamins do not prevent bronchopulmonary dysplasia in premature baboons exposed to prolonged hyperoxia: a pilot study. Pediatr Res 43: 719–726
Shenai JP 1999 Vitamin A supplementation in very low birth weight neonates: rationale and evidence. Pediatrics 104: 1369–1374
Tyson JE, Wright LL, Oh W, Kennedy KA, Mele L, Ehrenkranz RA, Stoll BJ, Lemons JA, Stevenson DK, Bauer CR, Korones SB, Fanaroff AA 1999 Vitamin A supplementation for extremely-low-birth-weight infants. National Institute of Child Health and Human Development Neonatal Research Network. N Engl J Med 340: 1962–1968
Kennedy KA, Stoll BJ, Ehrenkranz RA, Oh W, Wright LL, Stevenson DK, Lemons JA, Sowell A, Mele L, Tyson JE, Verter J 1997 Vitamin A to prevent bronchopulmonary dysplasia in very-low-birth-weight infants: has the dose been too low? The NICHD Neonatal Research Network. Early Hum Dev 49: 19–31
Berger TM, Frei B 1995 Pro- or antioxidant activity of vitamin C in preterm infants?. Arch Dis Child Fetal Neonatal Ed 72: F211–F212
Saldanha RL, Cepeda EE, Poland RL 1982 The effect of vitamin E prophylaxis on the incidence and severity of bronchopulmonary dysplasia. J Pediatr 101: 89–93
Watts JL, Milner R, Zipursky A, Paes B, Ling E, Gill G, Fletcher B, Rand C 1991 Failure of supplementation with vitamin E to prevent bronchopulmonary dysplasia in infants less than 1,500 g birth weight. Eur Respir J 4: 188–190
Johnson L, Bowen FW Jr, Abbasi S, Herrmann N, Weston M, Sacks L, Porat R, Stahl G, Peckham G, Delivoria-Papadopoulos M, Quinn G, Schaffer D 1985 Relationship of prolonged pharmacologic serum levels of vitamin E to incidence of sepsis and necrotizing enterocolitis in infants with birth weight 1,500 grams or less. Pediatrics 75: 619–638
Ahola T, Lapatto R, Raivio KO, Selander B, Stigson L, Jonsson B, Jonsbo F, Esberg G, Stovring S, Kjartansson S, Stiris T, Lossius K, Virkola K, Fellman V 2003 N-acetylcysteine does not prevent bronchopulmonary dysplasia in immature infants: a randomized controlled trial. J Pediatr 143: 713–719
Sandberg K, Fellman V, Stigson L, Thiringer K, Hjalmarson O 2004 N-acetylcysteine administration during the first week of life does not improve lung function in extremely low birth weight infants. Biol Neonate 86: 275–279
Davis JM, Parad RB, Michele T, Allred E, Price A, Rosenfeld W 2003 Pulmonary outcome at 1 year corrected age in premature infants treated at birth with recombinant human CuZn superoxide dismutase. Pediatrics 111: 469–476
Author information
Authors and Affiliations
Corresponding author
Additional information
The Role of Oxygen in Health and Disease - A Series of Reviews
This is the fourth article in the series of reviews focusing on the role that oxygen plays in health and disease. In this review Drs. Auten and Davis discuss reactive oxygen species (ROS) and signaling molecules for biological processes that contribute to adaptive or maladaptive molecular responses. The review also focuses on developmental molecular targets and therapeutic interventions such as antioxidant defenses and therapies.
Sherin U. Devaskar, M.D.
Editor in Chief
Rights and permissions
About this article
Cite this article
Auten, R., Davis, J. Oxygen Toxicity and Reactive Oxygen Species: The Devil Is in the Details. Pediatr Res 66, 121–127 (2009). https://doi.org/10.1203/PDR.0b013e3181a9eafb
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/PDR.0b013e3181a9eafb
This article is cited by
-
Advancing oral health: the antimicrobial power of inorganic nanoparticles
Journal of the Korean Ceramic Society (2024)
-
Exposure limits for indoor volatile substances concerning the general population: The role of population-based differences in sensory irritation of the eyes and airways for assessment factors
Archives of Toxicology (2024)
-
HS-GC–MS analysis of volatile organic compounds after hyperoxia-induced oxidative stress: a validation study
Intensive Care Medicine Experimental (2024)
-
Hyperoxia but not high tidal volume contributes to ventilator-induced lung injury in healthy mice
BMC Pulmonary Medicine (2023)
-
Cellular stress in the pathogenesis of nonalcoholic steatohepatitis and liver fibrosis
Nature Reviews Gastroenterology & Hepatology (2023)
