
Abstract
Inborn errors of urea synthesis lead to an accumulation of ammonia in blood and brain and result in high rates of mortality and neurodevelopmental disability. This study seeks to characterize the cognitive, adaptive, and emotional/behavioral functioning of children with urea cycle disorders (UCDs). These domains were measured through testing and parent questionnaires in 92 children with UCDs [33 neonatal onset (NO), 59 late onset (LO)]. Results indicate that children who present with NO have poorer outcome than those who present later in childhood. Approximately half of the children with NO performed in the range of intellectual disability (ID), including a substantial number (∼30%) who were severely impaired. In comparison, only a quarter of the LO group was in the range of ID. There is also evidence that the UCD group has difficulties in aspects of emotional/behavioral and executive skills domains. In conclusion, children with UCDs present with a wide spectrum of cognitive outcomes. Children with NO disease have a much higher likelihood of having an ID, which becomes even more evident with increasing age. However, even children with LO UCDs demonstrate evidence of neurocognitive and behavioral impairment, particularly in aspects of attention and executive functioning.
Similar content being viewed by others


Main
Urea cycle disorders (UCDs) comprise deficiencies in any of the six enzymes and two transport proteins involved in urea biosynthesis. The specific disorders are: n-acetylglutamate synthase deficiency, carbamyl phosphate synthetase I deficiency, ornithine transcarbamylase (OTC) deficiency, argininosuccinate synthetase (AS) deficiency (citrullinemia), argininosuccinate lyase (AL) deficiency (argininosuccinic aciduria), arginase deficiency (argininemia), hyperornithinemia, hyperammonemia, homocitrullinuria syndrome (or mitochondrial ornithine carrier deficiency-ORNT), and citrullinemia type II (mitochondrial aspartate/glutamate carrier deficiency-CITR). UCDs are inherited as autosomal recessive traits, except for OTC deficiency, which is X-linked. Because of the absence of nationwide newborn screening for these disorders, the true incidence of UCD is unknown. Based on case reports and data from referred patients, the estimated combined incidence for all UCDs has ranged from 1 in 8,200 to 1 in 30,000 (1,2).
Inborn errors of urea synthesis lead to an accumulation of ammonia in blood and brain and clinically present as recurrent episodes of hyperammonemia manifested by vomiting, lethargy, and coma. Infants with complete enzyme deficiencies (other than argininemia) commonly present in the newborn period with hyperammonemic coma. Despite aggressive treatment with hemodialysis, the mortality in the newborn period for proximal urea cycle defects (OTC deficiency and CPSI deficiency) has been reported to approach 50% (3). As reported in our previous studies, virtually all survivors have developmental disabilities that correlate with the number, severity, and duration of hyperammonemic episodes (4,5). Studies of children with neonatal UCDs have shown variable intellectual outcomes, based broadly on developmental quotients (DQs) or on traditional intelligence quotient (IQ) scores. In general, these studies have demonstrated a high percentage of the group (∼60–80%) with mental retardation (4,6). Patients with partial defects may present at any age with hyperammonemic episodes that approximately carry a 10% mortality rate and a significant risk for developmental disabilities (7). In addition, because OTC inheritance is X-linked, males and females are differentially affected (3,8,9), particularly because the females are heterozygous carriers (10,11). Males are more likely to present neonatally with more severe hyperammonemic episodes, whereas the clinical presentation of females varies, typically with a later symptom onset (8). However, even asymptomatic OTC-deficient heterozygotes have been reported to have cognitive deficits and are at risk for learning disabilities and attention deficit hyperactivity disorder (ADHD) (10,11). Recent imaging studies have also shed light on the brain-behavior correlates associated with hyperammonemia (12), suggesting that acute hyperammonemia selectively affects white matter.
The mainstay of pharmacological therapy (6,13,14) involves the use of alternative pathway therapy (phenylbutyrate-buphenyl) to scavenge nitrogen, combined with supplementation of the amino acids l-citrulline or l-arginine. This approach, although often life-saving, fails to prevent severe neurologic sequelae during a hyperammonemic crisis. The only known cure is liver transplantation, which also carries significant morbidity (15). Nutritional management is also important and involves restriction of dietary protein along with appropriate supplementation (16).
The mortality and morbidity rates currently cited for UCDs were obtained before Food and Drug Administration approval of more effective therapy using the ammonia scavenger sodium phenybutyrate (buphenyl) in 1996. In the early 1980s, our group reported the 5-y survival rate for a small prospective cohort of neonatal onset (NO) disease to be ∼25%, and the morbidity (e.g., intellectual disability (ID), seizure disorders, and cerebral palsy) to be virtually universal for infants rescued from hyperammonemic coma (4,14). For distal defects (AS and AL deficiency), the neonatal mortality rate was significantly lower, ∼10%, but morbidity remained high. It has been hypothesized that higher morbidity in children with these distal defects may be related to additional other factors outside of hyperammonemia (17). The increased access to blood ammonia testing at most hospitals, heightened index of suspicion for metabolic disorders, availability of molecular tools for diagnosis (18), availability of newborn screening for some of the distal disorders, and availability of standardized medical therapy since 1996 have likely improved both ascertainment and outcome.
In 2003, the National Institutes of Health funded 10 rare Diseases Clinical Research Centers, one focusing on UCD. A principal goal of these centers is to carry out a natural history study of the disorder. Our longitudinal study started in 2006. This article presents an interim analysis of select aspects of the baseline neuropsychological data of children enrolled in this study. Specifically, we present profiles of intellectual, adaptive, and emotional/behavioral functioning of children with UCDs and evaluate group differences in these functional areas among neonatal vs. late onset (LO) presentation and the most prevalent diagnoses. We also aim to better characterize the prevalence of both mild and more severe disability in these groups.
METHODS
Subjects.
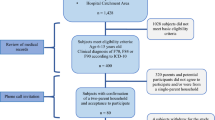
Individuals diagnosed with any one of the eight UCDs are being recruited for this study. The study was approved by all of the sites' IRBs and informed consent was obtained for all participants. Ninety-two participants are included in this interim analysis (age range at assessment, 5 mo to 16 y and 9 mo; mean, 7.2 y; SD, 4.7). Fifty-eight percent of the subjects were female, half of whom had partial OTC deficiency. Subjects were initially divided into groups based on age of disease onset. NO is defined as having had a clinical presentation within the first month of life. The LO group is defined as clinical onset beyond the first month of life or diagnosis based on family history, with confirmation by genetic testing. We also examined subjects based on history of hyperammonemia (either none or ≥1 episode). Children diagnosed by newborn screening who did not present with symptoms in the neonatal period were excluded from the current analysis, because of the limited number of subjects in this group.
Neuropsychological assessment.
Neuropsychological assessment was completed at or within 6 mo of the baseline visit for the overall study. The battery of tests administered varies with the participant's chronological and/or functional level, with each protocol designed to provide a comprehensive evaluation across functional domains. Those with IQ scores of 70 or below received an alternate, abbreviated battery. In developing the test battery for the study, a balance was struck between the need to use a standardized battery for all subjects and the reality that a significant subgroup will have severe ID such that it would not be possible to complete a standardized test battery appropriate to chronological age. For example, a 25-y-old adult subject may be functioning at a 2-y-old developmental level, making it necessary to use an alternative battery of tests to accurately document his/her true level of functioning.
Intellectual/developmental skills were assessed through individually administered tests including the Bayley Scales of Infant Development-Third Edition (Bayley-III, used for ages 5–35 mo) (19), the Wechsler Preschool and Primary Scale of Intelligence-Third Edition, (WPPSI-III, used for ages 3–5) (20), and the Wechsler Abbreviated Scales of Intelligence (WASI, used for ages 6–16) (21). Adaptive behavior, emotional/behavioral functioning, and everyday executive behavior were assessed though parent report questionnaires including the Adaptive Behavior Assessment System-Second Edition (ABAS-II, all ages) (22), Behavior Rating Inventory of Executive Function (BRIEF, ages 3–16) (23), and the Child Behavior Checklist (ages 3–16) (24), respectively.
In an attempt to include all individuals in the analysis of overall intelligence, estimated IQ scores were obtained for individuals with severe intellectual deficits not able to complete cognitive measures designed for their chronological age. When possible, these individuals were administered the Bayley-III and an age equivalence was obtained. This score was then converted into a DQ (test age/actual age) with a floor set at a score of 20. For individuals who were unable to complete the Bayley-III, we obtained caregiver ratings from the ABAS-II and assigned a score of 20. Finally, three individuals were not fluent in English and thus we included their Performance (nonverbal) IQ scores in the analysis. These alternative scores were only used in the analysis of overall intelligence.
Statistical analysis.
For the early childhood group, between groups analyses such as NO vs. LO groups or subjects with vs. without past hyperammonemic events were conducted using the Wilcoxon rank sum test, because of small number of samples. For the preschool/school age group, those analyses were conducted using two sample t test for each composite score from the intelligence (IQ) tests (WASI or WPPSI-III) and ABAS-II. In addition, because the scores on the IQ tests and the scores on the ABAS-II are highly correlated within each instrument, we used multivariate analysis of variance (MANOVA) to test if there was any difference between the NO and LO groups in these scores in each group. We also adjusted for age in years at the test since the age range is wide. For the OTC subjects in the preschool/school age group, between-group analyses was conducted for males vs. females using the Wilcoxon rank sum test.
For the overall analysis of intellectual ability, subjects in the preschool and school age group were divided into four categories based on intellectual level [full scale intelligence quotient (FSIQ)[rsbq] as follows: <50 = moderate to profound ID; 50–70 = mild ID; 70–85 = borderline/low average intelligence; >85 = broad range of average or above intelligence. Subjects in the OTC group were also examined by gender, as this is the only sex-linked disorder (X-linked recessive).
RESULTS
Most prevalent diagnoses in our samples were AL deficiency (18%), AS deficiency (20%), and OTC deficiency (47%). Majority of OTC patients were LO (81%) and a half of them were on buphenyl or citrulline (Table 1). There were no children with previously diagnosed mood disorders. Other psychiatric disorders, cerebral palsy, and autism spectrum disorders showed in low percentages, as opposed to learning disabilities and mental retardation/developmental disability (Table 2).
Early childhood group (n = 24).
In this group, most children performed at low average or above levels, with only a small percentage (8%, n = 2) with Bayley-III Composite scores in the mild range of impairment (SS <70). Both of these subjects had OTC with neonatal onset. No significant differences were found in Bayley-III scores or ABAS-II scores between NO and LO groups (p values ranged from 0.33 to 0.89); but there was a significant difference in Bayley-III Composite scores between subjects with and without hyperammonemic history (p = 0.0273). Table 3 provides a summary of the standard scores.
Preschool and school age group, ages 3–16 (n = 68).
In the NO group (n = 16), 50% had FSIQ in the range of ID. In the LO group (n = 52), in contrast, 25% were classified as having intellectual disabilities, with only a small percentage (4%) showing moderate to severe disabilities. Significant differences were found in both IQ (p = 0.0183 from MANOVA) and ABAS-II (p = 0.0055) scores between NO and LO groups; but, there were no differences between subjects with and without hyperammonemic history (p > 0.05). Table 4 summarizes the IQ and ABAS-II standard scores.
Intellectual data were also examined more closely in the three most common diagnoses (Fig. 1). For AL deficiency, none of the subjects presented with severe ID, although a substantial percentage (40%) had mild ID. For AS deficiency, 50% of the subjects presented with ID, with a substantial percentage (33%) presenting with moderate to severe disability.

Cognitive range across all subjects ages 3–16: NO vs. LO and by diagnosis. Estimated IQ scores for children unable to complete testing in their age range who were excluded from table 3 NO (n = 16), LO (n = 52), AL deficiency (n = 10; 40% NO), AS deficiency (n = 12; 58% NO), and OTC (n = 36; 8% NO). Average to above average (□); borderline/low ( ); mild ID (
); mild ID ( ); moderate/severe ID (▪).
); moderate/severe ID (▪).
In contrast, there were no significant differences between the groups in the emotional/behavioral and everyday executive skills domains (test results are not shown). However, there was evidence that the UCD group as a whole showed difficulty in these domains. Specifically, the UCD group demonstrated difficulties (ratings of ∼1–1 ½ standard deviations from the normal population) in several key areas including social difficulties, attention problems, and several metacognitive skills (initiation, planning/organization, working memory, and self-monitoring). Figure 2 illustrates the BRIEF profile for the school age group as a whole. The overall level of internalizing (e.g., anxiety and depression) and externalizing (e.g., aggression and conduct problems) behaviors was not as high, although the percentage of children with psychosocial problems in the UCD population seemed to be slightly greater than is expected in the normal population (Fig. 3).

UCD performance on the BRIEF for ages 6–16 relative to a normative population. INH, inhibit, SHF, shift, EMC, emotional control, INI, initiate, WKM, working memory, P/O, plan/organize, ORG, organization of materials, MON, monitor, BRI, behavioral regulation index, MI, metacognition index, GEC, global executive composite. UCD group (•); normative mean (solid line); 1 SD (dashed line).

UCD subjects ages 3–16: child behavior checklist. Clinically significant T ≥ 70 (▪); at risk T = 60–69 ( ); within normal limits (□).
); within normal limits (□).
OTC deficiency.
For OTC deficiency, 20% presented with ID and only a small percentage with severe ID (6%). Given the high number of females in this group (2:1 ratio), this group was further analyzed by gender. The male subjects were younger at the time of symptom onset (1.7 vs. 3.9 y) and similarly more likely to have early onset disease (41 vs. 4%). Although, no significant differences were found between males and females for any of the summary measures (p values all >0.10), the males had a higher prevalence of intellectual disabilities in their group (33 vs. 13%) when the lowest functioning (“untestable”) subjects (n = 2 male OTC subjects) were included in the count.
DISCUSSION
Consistent with previous research, our results suggest that children presenting with NO UCDs have poorer outcome than those who present later in childhood. With regard to basic intelligence, approximately half of the NO group was in the range of ID, including a substantial number (∼30%) who were severely impaired. In comparison, only about a quarter of the LO group was in the range of ID with only a very small number (<5%) with moderate to severe disability. However, it is important to note that although there are fewer children with severe ID in this group, only 65% of the LO group showed IQ scores in the “average” range (compared with 19% in the NO group). Thus, although the LO group is performing much better than the NO group as whole, a substantial number are functioning below age expectations. Similar results were obtained for adaptive skills in that children with NO had greater impairments in comparison to children with LO disease. Across both groups, adaptive skills were generally commensurate with intellectual abilities. In addition, there is some preliminary evidence from the questionnaire data and our clinical experience that even children with average intellectual abilities have indications of other neuropsychological deficits, which will be quantitatively summarized in future articles. This is consistent with the finding that the children in our group have a higher number of previously reported diagnoses of learning disabilities and ADHD.
Although the discrepancy between the NO and LO groups in this study is similar to those identified in previous studies, the overall level of disability across both groups is notably lower than has been reported previously. In studies of individuals with NO disease, the percentage with any level of ID was as high as 60–80% in previous studies (4,6), as opposed to the 50% noted in this study. Similarly, previous research has suggested a higher proportion of disability in children with partial UCDs. We previously reported that >50% of school age subjects with partial UCDs were in the range of ID (5).
The relatively lower rates of intellectual impairment in this study may be attributable to several factors. First, the overall sample size in this study is substantially larger than the sample sizes in previous studies, which generally reported on less than 25 patients. Additionally, it is possible that there have been improvements in functional outcome as a result of improved screening, recognition, and treatment protocols. Finally, much of the previous work has only examined individuals with severe forms of the disease, whereas this study includes a wider range, many with milder disease presentations. For example, almost one third of the overall subjects were females with OTC, who are known to typically have a milder form of the disease. Although there were not significant differences noted on any of the summary measures of intelligence, adaptive functioning, or behavioral/emotional functioning between the females and males with OTC, the percentage of males with intellectual disabilities was substantially higher overall (33 vs. 13%) when the lowest functioning (“untestable”) individuals are included in the numbers.
Closer examination of the three most prevalent diagnoses (OTC, AL, and AS deficiencies), found the overall rates of ID to be variable. The OTC group had the lowest percentage of individuals with intellectual disabilities (20%), which is consistent with the low numbers of individuals in our OTC group with NO disease (∼8%). In contrast, both the AL and AS deficiency groups had a moderate percentage of subjects (40–50%) with intellectual disabilities, which is consistent with a higher percentage of individuals in these groups with NO disease. Interestingly, the AL and AS groups had equal or lower percentages of individuals with a history of at least one hyperammonemic episode (67% in AD deficiency group, 40% in the AL group vs. 64% in the OTC group). This finding is consistent with previous articles that have suggested that there may be additional mechanisms contributing to the cognitive dysfunction in individuals with the distal defects (17) such as argininosuccinic aciduria or citrulline. It is also interesting to note that although the overall rates for any level of ID were similar in the AS and AL deficiency groups, the AS group had several individuals with more severe intellectual disabilities. On closer examination, the AS deficiency group had a greater percentage of individuals with NO and with a history of at least one previous hyperammonemic episode. Again, it may be possible that cognitive dysfunction in these individuals is related to a combination of factors, not just hyperammonemia.
Results of our analysis for children under age 3 yielded somewhat different results than the preschool and school age sample. Specifically, 8% of the NO group and none of the LO onset group presented with significant cognitive delay, which is much lower than the percentage of preschool and school age children with intellectual deficits. In addition, there were no significant differences between the groups in either developmental or adaptive functioning. However, the composite scores for both groups were between ½ and 1 SD below the normative mean in all areas, including cognitive development, language, motor skills, and adaptive functioning. Finally, there was an affect on IQ for having a history hyperammonemia. Overall, these results suggests that although most of the young children with UCD do not present with severe impairments, there is evidence that their development is not entirely normal and that there is some impact of hyperammonemia, even if it does not present in the neonatal period.
There may be several reasons why fewer children are presenting with cognitive impairments in the youngest age group. One possibility is related to the sensitivity of the testing. The majority of the children in this group were tested at either age 6 mo or 18 mo of age. The range of behaviors that can be measured are more limited in early infancy, particularly before age 1. In addition, previous research has demonstrated poor predictability of the Bayley Scales of Infant Development (Second Edition) for cognitive ability at school age, both in medically at risk and normal populations (25–27). However, it has also been noted that although the predictive ability of the Bayley is somewhat poor, it is better in children with “neurosensory impairments.” Thus, poor performance on the Bayley in children with UCDs who have significant neurologic impairments may indeed be a predictor of future cognitive difficulties in childhood.
A second possibility is that over time, children with UCD may demonstrate stagnation, slowed development, or even a decline in abilities related to their disease process, particularly if characterized by recurrent episodes of hyperammonemia. This would be consistent with previous studies (9), which demonstrated decline in cognitive scores over time in a group of females with OTC. Although we do not yet have enough longitudinal data to answer this question, we evaluated the mean scores across several age ranges to identify evidence supporting this hypothesis. There was some evidence suggesting that overall cognitive/intellectual scores in the cross-sectional analyses of children in the 0–3, 3–5, and 6–16 age bands decline over time, particularly in the NO group (i.e., cognitive composite = 86 for 0–3, FSIQ = 78 for ages 3–5, and 71 for ages 6–16). This does not necessarily suggest that children with UCD are losing skills, but that their developmental trajectory is slower than that of the normal population.
Although significant differences were observed for intelligence and adaptive skills between the two age of onset groups, no differences were noted on parent report of behavioral, emotional, and executive functioning. There was no evidence of clinically significant behavioral or emotional problems in the group as a whole. However, compared with normative data, there is a slightly greater proportion of children with UCDs who have behavioral and emotional difficulties, though the majority have only mild problems (16–17% with mild difficulties and 4% with clinically significant symptoms). This included both internalizing (e.g., anxiety, depression, and withdrawal) and externalizing (e.g., aggression and conduct issues) symptoms. These results are consistent with a recent meta-analysis of psychosocial problems in children with chronic illnesses, which suggested that these children are at a slightly elevated risk of psychosocial distress, although only a minority experience clinical symptomatology (28). In addition, the children in this cohort show other behavioral/emotional strengths, including a minimal percentage with previous diagnoses of autism spectrum disorders, mood disorders, and other psychiatric disorders.
There is even greater evidence to suggest that individuals with UCD have difficulties with several aspects of attention and executive functioning. Executive functioning refers to a set of skills important for goal directed behavior and includes skills in cognitive, behavioral, and emotional arenas. In comparison with the normative population, our sample of children with UCDs was reported to have problems with attention and select metacognitive skills. This included their ability to sustain working memory, plan and organize their approach to tasks, and monitor their mistakes. These findings are consistent with previous articles, which noted executive functioning weaknesses in a group of adult females who were heterozygous carriers of the OTC gene (11). These results support the notion that there is selective vulnerability of white matter in individuals with UCD that is associated with hyperammonemia and is also consistent with neuroimaging studies (12). It will be critical to more closely examine the pattern of neuropsychological strengths and weaknesses in this population beyond measures of overall intelligence and development.
In summary, children with UCDs present with a wide variety of cognitive outcomes. Children with NO disease have a much higher likelihood of having an ID, which becomes even more evident with increasing age. However, even children with LO and presumably more mild disease presentation demonstrate evidence of neurocognitive and behavioral difficulties, particularly in aspects of executive functioning. A goal of future analyses will be to examine the broader neuropsychological profile of this population and identify factors that may predict their outcome. Finally, we also plan to further examine different disease subtypes and the relationship between proximal vs. distal enzyme defects and neuropsychological functioning.
Abbreviations
- ADHD:
-
attention deficit hyperactivity disorder
- AL:
-
argininosuccinate lysase deficiency
- AS:
-
argininosuccinate synthetase deficiency (citrullinemia)
- FSIQ:
-
full scale intelligence quotient
- ID:
-
intellectual disability
- IQ:
-
intelligence quotient
- LO:
-
late onset
- NO:
-
neonatal onset
- OTC:
-
ornithine transcarbamylase
- UCD:
-
urea cycle disorder
References
Brusilow S, Maestri NE 1996 Urea cycle disorders: diagnosis, pathophysiology, therapy. In: Barness LA, DeVivo DC, Kaback MM, Morrow G, Oski FA, Rudolph AM (eds) Advances in Pediatrics, Vol 43. Mosby, Chicago, p 127
Nagata N, Matsuda I, Oyanagi K 1991 Estimated frequency of urea cycle enzymopathies in Japan. Am J Med Genet 39: 228–229
Maestri NE, Clissold D, Brusilow SW 1999 Neonatal onset ornithine transcarbamylase deficiency: a retrospective analysis. J Pediatr 134: 268–272
Msall M, Batshaw ML, Suss R, Brusilow SW, Mellits ED 1984 Neurologic outcome in children with inborn errors of urea synthesis. N Engl J Med 310: 1500–1505
Msall M, Monahan PS, Chapanis N, Batshaw ML 1988 Cognitive development in children with inborn errors of urea synthesis. Acta Paediatr Jpn 30: 435–441
Batshaw ML, Brusilow SW, Waber L, Blom W, Brubakk AM, Burton BK, Cann HM, Kerr D, Mamunes P, Matalon R, Myerberg D, Schafer IA 1982 Treatment of inborn errors of urea synthesis: activation of alternate pathways of waste nitrogen synthesis and excretion. N Engl J Med 306: 1387–1392
Batshaw ML, Msall M, Trojak J 1986 The risk of serious illness in carriers of ornithine transcarbamylase deficiency. J Pediatr 108: 236–241
Nicolaides P, Leibsch D, Dale N, Surtees R 2002 Neurological outcome of patients with ornithine carbamoyltransferase deficiency. Arch Dis Child 86: 54–56
Maestri NE, Brusilow SW, Clissold DB, Bassett SS 1996 Long-term treatment of girls with ornithine transcarbamylase deficiency. N Engl J Med 335: 855–859
Batshaw ML, Roan Y, Rosenberg LA, Brusilow SW 1980 Cerebral dysfunction in asymptomatic carriers of ornithine transcarbamylase deficiency. N Engl J Med 302: 482–485
Gyato K, Wray J, Huang ZJ, Yudkoff M, Batshaw ML 2004 Metabolic and neuropsychological phenotype in women heterozygous for ornithine transcarbamylase deficiency. Ann Neurol 55: 80–86
Gropman AL, Summar M, Leonard JV 2007 Neurological implications of urea cycle disorders. J Inherit Metab Dis 30: 865–879
Brusilow SW, Batshaw ML, Valle D 1979 New pathways of waste nitrogen excretion in inborn errors of urea synthesis. Lancet 2: 452–454
Batshaw ML, MacArthur RB, Tuchman M 2001 Alternative pathway therapy for urea cycle disorders: twenty years later. J Pediatr 138: S46–S55
Whitington PF, Alonso EM, Boyle JT, Molleston JP, Rosenthal P, Emond JC, Millis JM 1998 Liver transplantation for the treatment of urea cycle disorders. J Inherit Metab Dis 21: 112–118
Singh RH 2007 Nutritional management of patients with urea cycle disorders. J Inherit Metab Dis 30: 880–887
Maestri NE, Hauser ER, Bartholomew D, Brusilow SW 1991 Prospective treatment of urea cycle disorders. J Pediatr 119: 923–928
Tuchman M, Jaleel N, Morizono H, Sheehy L, Lynch MG 2002 Mutations and polymorphisms in the human ornithine transcarbamylase gene. Hum Mutat 19: 93–107
Bayley N 2006 The Bayley Scales of Infant and Toddler Development, 3rd Ed. Harcourt Assessment, Inc, San Antonio, TX
Wechsler D 2002 Wechsler Preschool and Primary Scale of Intelligence, 3rd Ed. The Psychological Corporation, San Antonio, TX,
Wechsler D 1999 The Wechsler Abbreviated Scale of Intelligence. The Psychological Corporation, San Antonio, TX
Harrison PL, Oakland T 2003 Adaptive Behavior Assessment System, 2nd Ed. Harcourt Assessment, Inc, San Antonio, TX
Gioia GA, Isquith PK, Guy SC, Kenworthy L 2000 Behavior Rating Inventory of Executive Function. Psychological Assessment Resources, Inc, Lutz, FL
Achenbach TM 1991 Manual for the Child Behavior Checklist/4-18 and 1991 Profile. University of Vermont, Burlington, VT
Hack M, Taylor HG, Drotar D, Schluchter M, Cartar L, Wilson-Costello D, Klein N, Friedman H, Mercuri-Minich N, Morrow M 2005 Poor predictive validity of the Bayley Scales of Infant Development for cognitive function of extremely low birth weight children at school age. Pediatrics 116: 333–341
Aylward GP 2004 Prediction of functioning from infancy to early childhood: implications for pediatric psychology. J Pediatr Psychol 29: 555–564
Illingworth RS, Birch LB 1959 The diagnosis of mental retardation in infancy: a follow-up study. Arch Dis Child 34: 269–273
Barlow JH, Ellard DR 2006 The psychosocial well-being of children with chronic disease, their parents, and siblings: an overview of the evidence base. Child Care Health Dev 32: 19–31
Author information
Authors and Affiliations
Corresponding author
Additional information
Supported by National Institute of Health: Rare Diseases CRC—Urea Cycle Disorders, 5U54RR019453-04, The Kettering Fund, and the O'Malley Family Foundation.
Rights and permissions
About this article
Cite this article
Krivitzky, L., Babikian, T., Lee, HS. et al. Intellectual, Adaptive, and Behavioral Functioning in Children With Urea Cycle Disorders. Pediatr Res 66, 96–101 (2009). https://doi.org/10.1203/PDR.0b013e3181a27a16
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/PDR.0b013e3181a27a16
This article is cited by
-
Using urine FTIR spectra to screen autism spectrum disorder
Scientific Reports (2023)
-
Relationship between longitudinal changes in neuropsychological outcome and disease biomarkers in urea cycle disorders
Pediatric Research (2023)
-
Sodium phenylbutyrate reduces repetitive self-grooming behavior and rescues social and cognitive deficits in mouse models of autism
Psychopharmacology (2021)
-
A retrospective review of outcomes in the treatment of hyperammonemia with renal replacement therapy due to inborn errors of metabolism
Pediatric Nephrology (2020)
-
Early liver transplantation in neonatal-onset and moderate urea cycle disorders may lead to normal neurodevelopment
Metabolic Brain Disease (2018)
