
Abstract
Early gestation dexamethasone (dex) administration is an ovine model of fetal programming associated with increased coronary reactivity to angiotensin II (Ang II). NADPH oxidase-dependent superoxide production plays an important role in both Ang II signaling and coronary disease. We sought to determine whether early gestation dex-exposure increases coronary reactivity to Ang II by enhancing endothelial NADPH oxidase-dependent superoxide production. Dex (0.28 mg/kg/d for 48 h) was administered to pregnant ewes at 27–28 d gestation. Dex-exposed and control offspring were studied at 4 mo of age. Coronary superoxide production was measured by lucigenin-enhanced chemiluminescence and dihydroethidium fluorescence. Coronary arteries from dex-exposed sheep had significantly enhanced vasoconstriction to Ang II, an effect abolished by either endothelial removal or preincubation with membrane-permeable superoxide dismutase and catalase. Ang II significantly increased endothelial superoxide production and NADPH oxidase activity in coronaries from dex-exposed offspring, but not controls. This programmed alteration in superoxide production was accentuated by PD123319 (AT2 antagonist), but abolished by losartan (AT1 antagonist). In conclusion, early gestation dex-exposure programs coronary reactivity to Ang II by enhancing Ang II-stimulated endothelial superoxide production. This programming effect may predispose to progressive coronary endothelial dysfunction and coronary artery disease.
Similar content being viewed by others

Main
An adverse intrauterine environment increases the risk of developing adult diseases, including hypertension, diabetes, and obesity (1). Animal models ranging from maternal undernutrition to uterine artery ligation or placental embolization have been used to induce metabolic syndrome in adult offspring (1). Mechanistically, these models impair fetal growth and increase fetal glucocorticoid exposure through down-regulation of placental 11β-hydroxysteroid dehydrogenase (2,3). To better evaluate the effects of fetal glucocorticoid exposure on cardiovascular function later in life, in the absence of confounding alterations in maternal health and fetal growth, intrauterine glucocorticoid exposure models of fetal programming were developed (4). Using sheep, a species with cardiovascular developmental trajectory relatively analogous to that of humans, Dodic et al. noted first trimester dexamethasone (dex) exposure elicits offspring hypertension by 4 mo of age, despite normal intrauterine and postnatal growth (5).
Using this dex-exposure model, we have demonstrated that programmed sheep have coronary artery-specific increases in both superoxide production and angiotensin II (Ang II) responsiveness without alteration in Ang II receptor expression (6,7). Supporting the relevance of these findings, a role for renin-angiotensin system activation in programmed cardiovascular disease has now been demonstrated in studies that bridge animal models and experimental interventions (5,7–11). Among its cardiovascular effects, Ang II is a prototypical agonist for vascular NADPH oxidase, an enzyme linked to progression of atherosclerotic plaques through increased endothelial superoxide production (12,13). Therefore, we hypothesized that early gestation dex-exposure increases postnatal coronary constriction to Ang II by enhancing endothelial NADPH oxidase-dependent superoxide production.
METHODS
Animal model.
The investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (National Institutes of Health Publication No. 85-23, revised 1996) and was approved by the University of Iowa Animal Care and Use Committee. Time-dated pregnant ewes were obtained from Iowa State University. At 27–28 d gestation (term 145 d), dex (0.28 mg/kg/d; Sicor Pharmaceuticals, Irvine, CA) was administered to the ewes by continuous i.v. infusion over 48 h. Dex-exposed lambs were pasture-raised in a fashion identical to that used for age-matched control lambs. At 4 mo of age, these lambs were killed with i.v. pentobarbital sodium (50 mg/kg; Abbott Laboratories, Abbott Park, IL).
Coronary reactivity.
Proximal left anterior descending coronary artery segments were immediately dissected from dex-exposed and control sheep (n = 7). Endothelium was removed from one arterial segment by rubbing. Rings were then mounted in wire myographs and equilibrated for 1 h, as previously described (7). After equilibration, arteries were constricted with KCl (120 mM) to normalize subsequent reactivity data. After washing, endothelium-intact rings were incubated for 10 min in either buffer alone, indomethacin (10−5 M), or a combination of polyethylene glycol-conjugated catalase (PEG-catalase) (250 units/mL) and PEG-superoxide dismutase (86 units/mL) to evaluate the role of prostanoids and reactive oxygen species in Ang II-mediated reactivity. Response was then measured to cumulative concentrations of Ang II (10−11–10−7 M every 2 min). After re-equilibration, each ring was constricted with the thromboxane A2 agonist U46619 before dilation with bradykinin (10−7 M) and then sodium nitroprusside (10−5 M) to evaluate endothelial and smooth muscle integrity, respectively.
Fluorescent in situ superoxide anion detection.
Immediately after harvest, distal left anterior descending coronary segments were incubated for 4 h in buffer with or without 10−7 M Ang II. The segments were then cryosectioned (30 μm) and incubated for 30 min at 37°C with dihydroethidium (2 μM), a fluorophore selective for superoxide anion (14). Fluorescence was detected with a Bio-Rad laser scanning confocal microscope (excitation 488 nm; detection 585 nm). Control and dex-exposed vessels were analyzed in parallel under identical laser settings, as previously described (6).
Chemiluminescence.
Distal left anterior descending coronary arteries were further sectioned into 3 mm rings and opened longitudinally to increase the exposure of the endothelial layer. Based on preliminary studies in isolated coronaries showing that Ang II stimulates an immediate burst of superoxide production followed by a sustained increase 3–4 h later and that this superoxide production is greater after incubation with 10−7 M Ang II than with 10−9 M Ang II (data not shown), coronary segments were incubated at 37°C for 4 h in buffer with or without Ang II (10−7 M). Basal superoxide anion production was measured by lucigenin (25 μM)-enhanced chemiluminescence after 5 min of dark adaptation (luminometer model FB12; Zylux) (6). NADPH oxidase-dependent superoxide production was then measured as the diphenylene-iodonium (10−4 M)-inhibitable chemiluminescence measured after the addition of the enzyme substrate NADPH (10−4 M). To evaluate the role of Ang II type 1 and type 2 receptors (AT1 and AT2) in Ang II-stimulated NADPH oxidase-dependent superoxide production, coronary segments from a second group of dex-exposed and control sheep (n = 7) were preincubated for 5 min in either buffer alone, PD123319 (AT2 antagonist; 10−5 M) or losartan (AT1 antagonist; 5 × 10−6M) before the 4-h Ang II incubation.
Chemicals.
Dihydroethidium was obtained from Molecular Probes (Eugene, OR). All other chemicals, unless indicated, were obtained from Sigma Chemical Co.—Aldrich (St. Louis, MO).
Data analysis.
All values are presented as mean ± SE. Statistical comparisons were performed by Student unpaired, two-tailed t tests or analysis of variance, as appropriate. A value of p < 0.05 was considered significant. If analysis of variance identified significant differences, pairwise comparisons were made using the Tukey test. All analyses were performed using SAS System 9 for Microsoft Windows (SAS Institute Inc., Cary, NC).
RESULTS
Coronary reactivity.
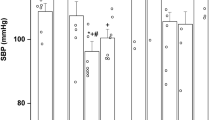
Vasoconstriction to 120 mM KCl and vasodilation to 10−7 M bradykinin were not significantly altered by dex-exposure (Table 1). Compared with intact rings, rubbed rings had similar vasoconstriction to KCl, but significantly decreased dilation to bradykinin (Table 1). All vessels dilated completely to sodium nitroprusside (data not shown). Early gestation dex-exposure led to significantly increased vasoconstriction to Ang II (Fig. 1A). Although indomethacin significantly enhanced contractile responses in both groups (p < 0.01), indomethacin preincubation did not eliminate the dex-induced increase in Ang II responsiveness (Fig. 1C). In contrast, the dex-enhanced Ang II contractility was eliminated by either endothelial rubbing (Fig. 1B) or preincubation with a combination of PEG-catalase and PEG-superoxide dismutase (Fig. 1D). Across all studies of Ang II-mediated vasoconstriction (Fig. 1), utilization of cumulative concentrations of Ang II without interposed vessel re-equilibration led to characteristic attenuation in constriction with prolonged Ang II exposure (tachyphylaxis).

Cumulative concentration-response curves for coronary vasoconstriction to angiotensin II in dexamethasone-exposed (▪) or control (○) lambs (n = 7). Responses were assessed in intact rings (A), rubbed rings (B), and intact rings after preincubation with either indomethacin (C) or a combination of PEG-superoxide dismutase and PEG-catalase (D). Values are displayed as means with vertical lines indicating SE. *Significant differences between dexamethasone infused and control lambs (p < 0.05).
Coronary superoxide production.
Dihydroethidium fluorescence was increased within coronary arteries from dex-exposed lambs (Fig. 2B, compared with control Fig. 2A). After incubation with Ang II, endothelial superoxide levels were further increased in early gestation dex-exposed lambs (Figs. 2D and F), but not control lambs (Figs. 2C and E). Previous dex-exposure was similarly associated with increased lucigenin-induced chemiluminescence (Fig. 3A) and increased Ang II-stimulated superoxide production (Fig. 3B). Although NADPH oxidase-dependent superoxide production was not altered after dex-exposure (Dex: 2022 ± 349 RLU/mg/s; Control: 1795 ± 296 RLU/mg/s), Ang II incubation increased NADPH-stimulated superoxide production only among coronaries obtained from dex-exposed lambs (Fig. 4). In the presence of the AT2 antagonist PD123319, previous dex-exposure led to significantly increased Ang II-stimulated superoxide production (Fig. 4; p < 0.05 vs. control). These Ang II-induced alterations in superoxide production were completely blocked by the AT1 antagonist losartan (Fig. 4).

Intracellular superoxide anion levels detected by dihydroethidium (DHE) fluorescence in control (left column) or early gestation dexamethasone-exposed (right column) coronary arteries after 4 h incubation in buffer alone (row 1, 40×) or buffer with 10−7 M angiotensin II (row 2, 40×; row 3, 200×). Scale bar = 100 μM.

Superoxide production by coronary artery segments was measured by lucigenin-enhanced chemiluminescence in relative light units (RLU) · mg dry weight−1 · s−1. Coronary segments from control (□) and dexamethasone-exposed (▪) lambs were incubated for 4 h in either buffer alone (A) or buffer with 10−7 M angiotensin II (B). *p < 0.05 versus control (n = 7).

NADPH oxidase-dependent superoxide production by coronary arteries from control (□) and dex-exposed (▪) lambs was measured by lucigenin-enhanced chemiluminescence after incubation for 4 h in 10−7 M angiotensin II (Ang II) with and without preincubation with PD123319 (AT2 antagonist) or losartan (AT1 antagonist). Values are normalized by the amount of superoxide produced after incubation in buffer alone. *Significant difference between dexamethasone infused and control lambs (p < 0.05).
DISCUSSION
Epidemiologic studies have identified a range of environmental factors that predispose to coronary heart disease (15). Low birth weight, a marker of an adverse intrauterine environment, was first identified as an independent risk factor for coronary mortality 2 decades ago (16). Animal models of fetal programming were quickly developed to identify the causes of programmed cardiovascular disease. Among these models, dex administration early in sheep pregnancy has uniquely allowed comprehensive evaluation of offspring coronary artery physiology in the absence of confounding effects on fetal or postnatal growth. Using this model, we have previously demonstrated coronary-specific increases in AT1-dependent vasoconstriction (7). Importantly, this physiologic alteration occurred in the absence of alteration in AT1 protein expression, raising the possibility that down-stream signaling pathways were permanently programmed by the early gestation steroid exposure. We now show that this prenatally induced alteration in coronary Ang II responsiveness is mechanistically linked to exaggerated endothelial superoxide production and further show that Ang II can exacerbate postnatal coronary dysfunction through AT1-mediated enhancement in NADPH oxidase-dependent superoxide production.
The interrelationships between Ang II, NADPH oxidase activity, and coronary artery dysfunction have been a source of intense investigation. Studies of human coronary artery sections have demonstrated up-regulation of both tissue renin-angiotensin systems and NADPH oxidase subunits in atherosclerotic vessels (13,17,18). Further studies have shown that even in the absence of atherosclerosis, endothelial NADPH oxidase is the major source of coronary superoxide (19), and infusion of Ang II increases vascular superoxide production by NADPH oxidase via direct stimulation of AT1 receptors (20,21).
Just as superoxide production plays a central role in the inception and progression of coronary dysfunction and Ang II-mediated hypertension, superoxide-mediated endothelial dysfunction has been shown to play a key role in the pathogenesis of both glucocorticoid-induced and genetically determined hypertension (12,22,23). With many models of fetal programming associated with both exaggerated glucocorticoid exposure and altered renin-angiotensin system activity (1), investigators have begun evaluating the role of Ang II-stimulated reactive oxygen species production in programmed hypertension. Using a rat undernutrition model, Franco et al. demonstrated that hypertensive male offspring had increased mesenteric Ang II concentrations and endothelial dysfunction as a consequence of enhanced AT1 receptor-mediated superoxide production (9). Similarly, male offspring of dams provided with a low protein diet through pregnancy displayed exaggerated carotid vasoconstriction to Ang II due to enhanced AT1 receptor-mediated NADPH oxidase-dependent superoxide production (10).
Our study supports and extends those findings by showing that coronary arteries from 4-mo-old male and female sheep previously exposed to an adverse intrauterine environment have enhanced vasoconstriction to Ang II due to a programmed increase in endothelial superoxide production. Although the referenced studies (9,10) only evaluated male offspring and some rodent studies have demonstrated sex differences in programmed hypertension (24,25), a majority of such studies ultimately reveal programmed hypertension in both genders (26–32). This is consistent with human epidemiologic studies revealing similar relations between birth weight and cardiovascular death or hypertension in men and women (33–35). Although we are not aware of any fetal programming models that have demonstrated sex-specific programming of coronary physiology, our inability to perform meaningful subgroup analysis within the context of the present study may hamper the generalization of our results to other model systems.
The endothelial origin of the programmed coronary dysfunction was demonstrated both by wire myography and confocal microscopy. Although the wire myography studies revealed an important role for vasodilatory prostanoids in the regulation of coronary tone among both control and dex-exposed sheep, it was preincubation with reactive oxygen species scavengers not indomethacin that blocked the development of the heightened Ang II-mediated vasoconstriction. Although preincubation with both membrane permeable superoxide dismutase and catalase confirmed a role for reactive oxygen species in programmed coronary dysfunction, use of the two compounds simultaneously limited our ability to explore, by wire myography, the reactive oxygen species predominately affected. We chose to use these agents in combination, rather than using superoxide dismutase in isolation, to avoid accumulation of hydrogen peroxide, a potent coronary vasodilator (36). Subsequent studies were then designed with probes selective for superoxide to clarify our vascular reactivity findings. The results seen by lucigenin-enhanced chemiluminescence and dihydroethidium fluorescence provide converging lines of evidence that early gestation dex induces coronary superoxide production.
Although we did not fully evaluate alternative sources of superoxide, including the mitochondrial electron transport chain, xanthine oxidase, cyclooxygenase, and uncoupled nitric oxide synthase, NADPH oxidases are considered the major sources of vascular superoxide (37). This is supported by the nearly 100-fold increase in coronary superoxide production seen after addition of the NADPH oxidase substrate (NADPH), and the nearly complete loss of superoxide production after subsequent addition of the flavoprotein inhibitor diphenyleneiodonium. Finally, although we did not evaluate endogenous antioxidant activity within the context of these studies, our conclusion that the primary alteration is enhanced superoxide production, rather than impaired reactive oxygen species (ROS) scavenging, is supported by the chemiluminescence patterns, as well as the growing body of literature now revealing potent regulatory effects of Ang II on NADPH oxidase expression (20,21).
Perspectives.
The present study provides novel information regarding glucocorticoid-programmed alterations in coronary artery physiology in the absence of fetal or postnatal growth restriction. Glucocorticoid exposure seems to be a final common pathway whereby a variety of adverse environmental exposures, from maternal undernutrition to psychological stress, prepare the fetus for postnatal survival, potentially at the expense of adult cardiovascular health. If the heightened Ang II responsiveness and superoxide production seen herein is similarly programmed in humans with a genetic predisposition to coronary disease, accelerated atherosclerosis and cardiovascular mortality may follow. Further studies in prehypertensive newborn lambs are necessary to ascertain whether the dex-induced ANGII-dependent superoxide production we have demonstrated is a primary phenotype or a consequence of hypertension-induced endothelial dysfunction. Regardless of its antecedents, the biologic ramifications of this environmentally programming coronary physiology would presumably be most harmful if it occurs in conjunction with increased renin angiotensin system activity, as may occur during the evolution of reno-vascular hypertension.
Abbreviations
- Ang:
-
angiotensin
- AT1:
-
angiotensin II type 1 receptor
- AT2:
-
angiotensin II type 2 receptor
- Dex:
-
dexamethasone
- PEG:
-
polyethylene glycol
- ROS:
-
reactive oxygen species
References
McMillen IC, Robinson JS 2005 Developmental origins of the metabolic syndrome: prediction, plasticity, and programming. Physiol Rev 85: 571–633
Langley-Evans SC, Phillips GJ, Benediktsson R, Gardner DS, Edwards CR, Jackson AA, Seckl JR 1996 Protein intake in pregnancy, placental glucocorticoid metabolism and the programming of hypertension in the rat. Placenta 17: 169–172
Homan A, Guan H, Hardy DB, Gratton RJ, Yang K 2006 Hypoxia blocks 11beta-hydroxysteroid dehydrogenase type 2 induction in human trophoblast cells during differentiation by a time-dependent mechanism that involves both translation and transcription. Placenta 27: 832–840
Benediktsson R, Lindsay RS, Noble J, Seckl JR, Edwards CR 1993 Glucocorticoid exposure in utero: new model for adult hypertension. Lancet 341: 339–341
Dodic M, May CN, Wintour EM, Coghlan JP 1998 An early prenatal exposure to excess glucocorticoid leads to hypertensive offspring in sheep. Clin Sci 94: 149–155
Roghair RD, Miller FJ Jr, Scholz TD, Lamb FS, Segar JL 2007 Endothelial superoxide production is altered in sheep programmed by early gestation dexamethasone exposure. Neonatology 93: 19–27
Roghair RD, Lamb FS, Miller FJ Jr, Scholz TD, Segar JL 2005 Early gestation dexamethasone programs enhanced postnatal ovine coronary artery vascular reactivity. Am J Physiol Regul Integr Comp Physiol 288: R46–R53
Pladys P, Sennlaub F, Brault S, Checchin D, Lahaie I, Lê NL, Bibeau K, Cambonie G, Abran D, Brochu M, Thibault G, Hardy P, Chemtob S, Nuyt AM 2005 Microvascular rarefaction and decreased angiogenesis in rats with fetal programming of hypertension associated with exposure to a low-protein diet in utero. Am J Physiol Regul Integr Comp Physiol 289: R1580–R1588
Franco Mdo C, Akamine EH, Di Marco GS, Casarini DE, Fortes ZB, Tostes RC, Carvalho MH, Nigro D 2003 NADPH oxidase and enhanced superoxide generation in intrauterine undernourished rats: involvement of the renin-angiotensin system. Cardiovasc Res 59: 767–775
Yzydorczyk C, Gobeil F Jr, Cambonie G, Lahaie I, Lê NL, Samarani S, Ahmad A, Lavoie JC, Oligny LL, Pladys P, Hardy P, Nuyt AM 2006 Exaggerated vasomotor response to ANG II in rats with fetal programming of hypertension associated with exposure to a low-protein diet during gestation. Am J Physiol Regul Integr Comp Physiol 291: R1060–R1068
Singh RR, Cullen-McEwen LA, Kett MM, Boon WM, Dowling J, Bertram JF, Moritz KM 2007 Prenatal corticosterone exposure results in altered AT1/AT2, nephron deficit and hypertension in the rat offspring. J Physiol 579: 503–513
Griendling KK, Sorescu D, Ushio-Fukai M 2000 NADPH oxidase: role in cardiovascular biology and disease. Circ Res 86: 494–501
Nickenig G, Harrison DG 2002 The AT1-type angiotensin receptor in oxidative stress and atherogenesis. I. Oxidative stress and atherogenesis. Circulation 105: 393–396
Walrand S, Valeix S, Rodriguez C, Ligot P, Chassagne J, Vasson M 2003 Flow cytometry study of polymorphonuclear neutrophil oxidative burst: a comparison of three fluorescent probes. Clin Chim Acta 331: 103–110
Rosamond W, Flegal K, Friday G, Go A, Greenlund K, Haase N, Ho M, Howard V, Kissela B, Kittner S, Lloyd-Jones D, McDermott M, Meigs J, Moy C, Nichol G, O'Donnell CJ, Roger V, Rumsfeld J, Sorlie P, Steinberger J, Thom T, Wasserthiel-Smoller S, Hong Y, American Heart Association Statistics Committee Stroke Statistics Subcommittee 2007 Heart disease and stroke statistics—2007 update: a report from the American Statistics Committee and Stroke Statistics Subcommittee. Circulation 115: e69–e171
Barker DJ, Winter PD, Osmond C, Margetts B, Simmonds SJ 1989 Weight in infancy and death from ischemic heart disease. Lancet 2: 577–580
Schmidt-Ott KM, Kagiyama S, Phillips MI 2000 The multiple actions of angiotensin II in atherosclerosis. Regul Pept 93: 65–77
Azumi H, Inoue N, Takeshita S, Rikitake Y, Kawashima S, Hayashi Y, Itoh H, Yokoyama M 1999 Expression of NADH/NADPH oxidase p22phox in human coronary arteries. Circulation 100: 1494–1498
Mohazzab KM, Kaminski PM, Wolin MS 1994 NADH oxidoreductase is a major source of superoxide anion in bovine coronary artery endothelium. Am J Physiol 266: H2568–H2572
Rajagopalan S, Kurz S, Münzel T, Tarpey M, Freeman BA, Griendling KK, Harrison DG 1996 Angiotensin II-mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NADPH oxidase activation. Contribution to alterations of vasomotor tone. J Clin Invest 97: 1916–1923
Zhang C, Hein TW, Wang W, Kuo L 2003 Divergent roles of angiotensin II AT1 and AT2 receptors in modulating coronary microvascular function. Circ Res 92: 322–329
Zhang Y, Croft KD, Mori TA, Schyvens CG, McKenzie KU, Whitworth JA 2004 The antioxidant tempol prevents and partially reverses dexamethasone-induced hypertension in the rat. Am J Hypertens 17: 260–265
Nakazono K, Watanabe N, Matsuno K, Sasaki J, Sato T, Inoue M 1991 Does superoxide underlie the pathogenesis of hypertension?. Proc Natl Acad Sci USA 88: 10045–10048
Langley-Evans SC, Welham SJ, Sherman RC, Jackson AA 1996 Weanling rats exposed to maternal low-protein diets during discrete periods of gestation exhibit differing severity of hypertension. Clin Sci 91: 607–615
Roghair RD, Aldape G 2007 Naturally occurring perinatal growth restriction in mice programs cardiovascular and endocrine function in a sex- and strain-dependent manner. Pediatr Res 62: 399–404
McMullen S, Langley-Evans SC 2005 Maternal low-protein diet in rat pregnancy programs blood pressure through sex-specific mechanisms. Am J Physiol Regul Integr Comp Physiol 288: R85–R90
Musha Y, Itoh S, Hanson MA, Kinoshita K 2006 Does estrogen affect the development of abnormal vascular function in offspring of rats fed a low-protein diet in pregnancy?. Pediatr Res 59: 784–789
Ozaki T, Nishina H, Hanson MA, Poston L 2001 Dietary restriction in pregnant rats causes gender-related hypertension and vascular dysfunction in offspring. J Physiol 530: 141–152
Sugden MC, Langdown ML, Munns MJ, Holness MJ 2001 Maternal glucocorticoid treatment modulates placental leptin and leptin receptor expression and materno-fetal leptin physiology during late pregnancy, and elicits hypertension associated with hyperleptinemia in the early-growth-retarded adult offspring. Eur J Endocrinol 145: 529–539
Vickers MH, Breier BH, Cutfield WS, Hofman PL, Gluckman PD 2000 Fetal origins of hyperphagia, obesity, and hypertension and postnatal amplification by hypercaloric nutrition. Am J Physiol Endocrinol Metab 279: E83–E87
Woods LL, Ingelfinger JR, Nyengaard JR, Rasch R 2001 Maternal protein restriction suppresses the newborn renin-angiotensin system and programs adult hypertension in rats. Pediatr Res 49: 460–467
Woods LL, Weeks DA 2004 Naturally occurring intrauterine growth retardation and adult blood pressure in rats. Pediatr Res 56: 763–767
Davies AA, Smith GD, May MT, Ben-Shlomo Y 2006 Association between birth weight and blood pressure is robust, amplifies with age, and may be underestimated. Hypertension 48: 431–436
Eriksson JG, Forsen TJ, Kajantie E, Osmond C, Barker DJ 2007 Childhood growth and hypertension in later life. Hypertension 49: 1415–1421
Osmond C, Barker DJ, Winter PD, Fall CH, Simmonds SJ 1993 Early growth and death from cardiovascular disease in women. BMJ 307: 1519–1524
Sato A, Sakuma I, Gutterman DD 2003 Mechanism of dilation to reactive oxygen species in human coronary arterioles. Am J Physiol Heart Circ Physiol 285: H2345–H2354
Li JM, Shah AM 2004 Endothelial cell superoxide generation: regulation and relevance for cardiovascular pathophysiology. Am J Physiol Regul Integr Comp Physiol 287: R1014–R1030
Author information
Authors and Affiliations
Corresponding author
Additional information
This study was supported by NIH grants ES-012268 (JLS), HL-04495 (TDS), HL-62483 (FSL), and HD-050359 (RDR).
Rights and permissions
About this article
Cite this article
Roghair, R., Miller, F., Scholz, T. et al. Coronary Constriction to Angiotensin II Is Enhanced by Endothelial Superoxide Production in Sheep Programmed by Dexamethasone. Pediatr Res 63, 370–374 (2008). https://doi.org/10.1203/PDR.0b013e3181659bfa
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/PDR.0b013e3181659bfa
