
Abstract
An imbalance in apoptosis or survival of immune cells plays an essential role in the pathophysiology of sepsis. Phagocytosis-induced cell death (PICD) is a common result of the pathogen-host cell interaction mediated by reactive oxygen species (ROS). Neonatal sepsis is frequently characterized by hyperinflammation. Cord blood monocytes (CBMO) are equivalent to monocytes of adults [peripheral blood monocytes (PBMO)], both in terms of phagocytosis and killing of Escherichia coli. We investigated whether CBMO are less sensitive toward PICD compared with PBMO. Monocytes were infected with green fluorescent protein (GFP)-labeled E. coli. Phagocytic activity, cell-count, Annexin V staining, hypoploid DNA content, CD95 and CD95L expression, and caspase-8 and -9 activities were analyzed by flow cytometry, ROS production by chemiluminescence, and CD95L mRNA expression by reverse-transcriptase polymerase chain reaction. With equal phagocytic activity and ROS production, PBMO cell count was decreased by 82 ± 6% versus 28 ± 8% for CBMO after infection. Annexin V binding was enhanced fivefold on PBMO; 56 ± 15% of PBMO showed a hypodiploid DNA content compared with 9 ± 6% of CBMO. Caspases CD95L and CD95L mRNA were up-regulated in PBMO. Our results indicate that CBMO are less sensitive toward E. coli-mediated PICD than PBMO. Modifying monocyte apoptosis may be a target for future interventions in sepsis.
Similar content being viewed by others


Main
Sepsis is a major health problem predominantly affecting preterm infants and is increasing in prevalence (1). Recent research has lead to defining the key role of apoptosis in the immune dysfunction that characterizes sepsis (reviewed in 2). Deletion of effector cells by apoptosis and induction of anergy in surviving cells by uptake of apoptotic particles has a profound impact on outcome and postseptic immune reactions. Autopsies of neonatal (3), pediatric (4), and adult septic patients revealed extensive apoptosis of lymphocytes and dendritic cells (5–9).
Apoptosis of neutrophils and monocytes after phagocytosis of bacteria (reviewed in 10) is called phagocytosis-induced cell death (PICD). Generation of reactive oxygen species (ROS) upon phagocytosis is thought to be one of the apoptosis-inducing events (11). An imbalance between survival and apoptosis would either lead to immune paralysis caused by effector cell loss or by an exaggerated, prolonged inflammatory reaction. Monocyte PICD may play a role in limiting inflammatory reactions and tissue destruction.
Clinically defined neonatal sepsis is accompanied by an excessive inflammatory response in which proinflammatory cytokines mediate severe cell and organ injury (12). Recent work has demonstrated diminished apoptosis in neonatal neutrophils compared with cells from adults (13,14); neonatal monocytes have not yet been investigated. We have previously shown that monocytes from cord blood (CBMO) and peripheral blood of healthy adults (PBMO) are equivalent in phagocytosis and degradation of green fluorescent protein (GFP)-labeled Escherichia coli (15). We therefore tested the hypothesis that E. coli-mediated PICD in CBMO would be reduced compared with PBMO.
MATERIALS AND METHODS
Patients.
The study protocol was approved by the Ethics Committee of Tuebingen University Hospital. All mothers gave written consent before they went into labor. Randomly selected, unrelated adults donated blood and served as controls. All term neonates were delivered spontaneously and did not exhibit signs of infection, as defined by clinical status, white blood cell count, and C-reactive protein. Mothers with amnion infection and prolonged labor were excluded. Umbilical cord blood was placed in heparin-coated tubes (4 IE/mL blood) immediately after cord ligation.
Reagents.
Antibodies to CD14 (MΦP9) and immunoglobulin (Ig)-matched controls (IgG1, IgG2b) were from BD Biosciences (Heidelberg, Germany), anti-CD95 (DX2) from Pharmingen/BD Biosciences, anti-CD95L (MHCD95L04) from Caltag/Invitrogen (Karlsruhe, Germany), Annexin V-PE from Miltenyi Biotec (Berglisch-Gladbach, Germany), and Fix-and-Perm-Solution from BD Biosciences. 4′,6-Diamidino-2-phenylindole-dihydrochloride (DAPI) was from Merck (Darmstadt, Germany). Propidium iodide (PI), staurosporine, isopropyl-β-D-thiogalactopyranosid, 5-amino-2,3,-dihydro-1,4,-phthalazinedione (luminol), and antibiotics were purchased from Sigma Chemical Co. (Munich, Germany) and mitomycin from Medac (Hamburg, Germany).
Cell cultures.
Peripheral blood and cord blood mononuclear cells (PBMC and CBMC) were isolated by density gradient centrifugation (Biochrom AG, Berlin, Germany). Washed cells were resuspended in VLE RPMI 1640 (Biochrom) containing 10% heat-inactivated fetal calf serum (FCS, Biochrom). Cells were counted in an ultraplane Neubauer hemocytometer, placed at 2 × 106 cells/mL in flat-bottom 24-well cell culture plates (Costar, Bodenheim, Germany), and incubated at 37°C.
Purification of monocytes.
Monocytes were separated by negative selection using the magnetic cell sorting monocyte isolation kit II (Miltenyi Biotec) according to the manufacturer's instructions. The purity of the resulting population was greater than 92% CD14 positive cells, as detected by fluorescence-activated cell sorting (FACS).
Bacterial culture.
E. coli DH5α, an encapsulated K12 laboratory strain, carrying the green fluorescent protein (gfp)-mut2 gene (16), was used for phagocytosis. Bacteria were freshly grown in Lennox-L-Broth-medium (Invitrogen) until early logarithmic growth, resuspended in phosphate-buffered saline (PBS), and used immediately.
Phagocytosis assay.
The phagocytosis assay was performed as previously described (15). Infection was performed for 60 min at a multiplicity of infection (MOI) of 1:50, if not otherwise indicated. MOI 1:5 was achieved by dilution with PBS. Actin-dependent phagocytosis was blocked by cytochalasin D (Cyto D, Sigma Chemical Co.; 10 μg/mL, 30 min before infection), resulting in a decrease of phagocytosis to less than 5% (17). For analysis of postphagocytic reactions, washed cells were cultured in RPMI supplemented with 10% FCS and gentamicin (Sigma Chemical Co.; 200 μg/mL).
Measurement of ROS production.
ROS production was measured by luminol-enhanced chemiluminescence as described by Gerber (18). Chemiluminescence assay was performed on a Biolumat (LB 9505; Berthold, Wildbad, Germany). PBMC or CBMC containing 1 × 105 monocytes were resuspended in 100 μL PBS supplemented with 5% glucose and luminol (200 μM) and preincubated for 30 min at 37°C in the absence or presence of Cyto D (10 μg/mL). Stimulation was started by adding E. coli. Reaction was monitored for 180 min at 37°C.
Enumeration of monocytes.
Monocytes were counted by flow cytometry using stem count beads (Beckman/Coulter, Krefeld, Germany). Cells were resuspended and carefully assessed by microscopy to ensure that monocytes were not adherent to the culture plates. A 100 μL cell suspension was mixed with 20 μL stem count beads (994/μL) and analyzed immediately. Cells were gated by forward versus side scatter, and beads were gated by fluorescence as a high-fluorescent, double-positive population. On the basis of the number of events in the bead region and a known bead concentration, absolute cell numbers were determined.
Annexin V staining.
Annexin V staining was assessed according to Brown and Savill (19). Cells were washed twice and resuspended in 50 μL in Annexin-buffer (0.02% sodiumazide in HEPES-buffer, pH 7.4), with 10 μL Annexin V-PE for 15 min, washed, and analyzed immediately.
Nuclear fragmentation.
Monocytes were stained with DAPI (1 μg/mL; Merck), centrifuged onto class slides with a Cytospin-16A-centrifuge (Hettich, Tuttlingen, Germany, 300 × g, 5 min), mounted in Fluoprep-mounting-medium (bioMérieux, Marcy lÉtoile, France), and analyzed with an Axioplan-2 microscope (Carl Zeiss, Jena, Germany) with the help of Isis-imaging software (MetaSytems, Altlussheim, Germany).
DNA fragmentation.
DNA fragmentation was assessed according to Nicoletti et al. (20). Washed cells were slowly resuspended in 2 mL of −20°C ethanol 70% with continuous vortexing and stored for 4 h at -20°C. Cells were washed twice, resuspended in 50 μL PBS containing 13 units RNase (DNase free; Sigma Chemical Co.), and incubated for 15 min at 37°C; 180 μL of PI (70 μg/mL) was added and incubated for 20 min, and analysis was performed immediately.
Caspase-8 and -9 detection.
Caspases were detected by Caspase-detection Kit 8 and 9 (Calbiochem/Merck, Darmstadt, Germany). Washed cells at 2.5 × 105 were mixed with 1 μL staining solution and incubated for 30 min at 4°C in the dark. Cells were washed twice with 500 μL washing buffer, centrifuged, resuspended in PBS, and analyzed. When indicated, caspase-inhibitor zVAD-fmk (50 μM; Calbiochem) was added to cell culture 1 h before infection.
Intracellular Cd95l stain.
After removal of bacteria, cells were washed with buffer [PBS with 0.1% bovine serum albumin (BSA; Sigma Chemical Co.)] and 0.1% sodium azide (Sigma Chemical Co.). One hundred microliters of Fix-and-Perm-Solution (BD Biosciences) were added for 20 min. Cells were washed [PBS with 0.5% BSA, 0.1% Saponin (Sigma Chemical Co.), 0.02% sodium azide], stained with anti CD95L or isotype control (Caltag/Invitrogen) for 30 min, washed twice, and analyzed.
CD95l MRNA quantification.
RNA extraction, cDNA synthesis, and real-time polymerase chain reaction (PCR) were performed as previously described (21). Primer-sequences were atc-atc-ttt-gga-gaa-gca-aat (primer 1), ggg-ata-ctt-aga-gtt-cct-cat-gtag (primer 2), agc-cca-gtt-tca-ttg-atc-aca-ag (probe 1), cca-ccc-ttc-tta-tac-ttc-act-cca-gaa (probe 2). Concentrations of target DNA were calculated to compare the cycle numbers of the log-linear phase of the samples with the cycle numbers of the external standards. All results were normalized against the expression of the housekeeping gene 5-aminolevulinate-synthase (ALAS).
Statistical analysis.
Results are expressed as mean ± SD. Mean fluorescence intensities (MFI) were determined and nonspecific background staining subtracted. Statistical analysis was performed using the decadic logarithm of the values of CD14, CD95L, Annexin V, and PI for t test (Sigmaplot 2000 software for Windows, SPSS, Chicago, IL). Values of p < 0.05 (adjusted according to Bonferroni-Holm for multiple group comparisons) were considered significant. Comparisons between means of caspase intensities were performed using the Mann-Whitney test. Analyses were performed with statistical software (Statistical Package for the Social Sciences, release 12.0 for Windows, SPSS, Chicago, IL).
RESULTS
Diminished PICD in CBMO.
Mononuclear cells were incubated with GFP-labeled E. coli, and extracellular bacteria were removed after 1 h (Tables 1 and 2 and Fig. 1). Phagocytic index, capacity, and ROS production were determined. Confirming earlier results, no differences were found between CBMO and PBMO (p > 0.05) Tables 1, upper, and 2 (15,22).

GFP-labeled E. coli-induced monocyte cell death. (A) PBMC (upper) and CBMC (lower) were infected with GFP-labeled E. coli for 1 h. Extracellular bacteria were removed and cells analyzed by way of forward and sideward scatter (FSC, SSC) after 24 h. One representative experiment. Circles represent the typical monocyte gates. (B) Relative numbers of monocytes (PBMO, ▪; CBMO, □) of living cells (n = 7, *p < 0.05 vs CBMO, §p < 0.05 vs infected PBMO). Cytochalasin D (Cyto D) was added 30 min before infection to block phagocytosis. (C) Cell counts of purified monocytes (PBMO, ▪; CBMO, □) after 24 h (n = 5; *p < 0.05 vs CBMO). Monocytes were either infected immediately after isolation (d 0) or after 48 h of preincubation (d 2).
To analyze postphagocytic survival, PBMC and CBMC were cultured for 24 h and enumerated. Size and granularity were determined by FACS, showing cell loss within the typical monocyte gate (demonstrated by circle), which was more pronounced in PBMC (Fig. 1A, upper) than CBMC (Fig. 1A, lower). Twenty-four h after E. coli infection, PBMO versus CBMO were diminished by 64 ± 9% versus 19 ± 6% (MOI 1:5, p < 0.05) and by 82 ± 6% versus 28 ± 8% (MOI 1:50, p < 0.05) (Fig. 1B), as quantified by FACS. In noninfected controls, the number of CBMO was smaller compared with PBMO (22 ± 6% vs 10 ± 4%; p < 0.05).
Monocyte death occurred when cells of nonmonocytic origin were present (Fig. 1B) or absent (Fig. 1C). Monocytes were negatively purified, infected with E. coli for 1 h, and extracellular bacteria were eliminated. After 24 h in tissue culture, PBMO versus CBMO were diminished by 56 ± 15% versus 31 ± 13% (MOI 1:5) and 73 ± 13% versus 24 ± 12% (MOI 1:50, all p < 0.05) (Fig. 1C). As described earlier, monocyte CD14 expression was down-regulated after bacterial challenge (15), and therefore this parameter was not used for quantification.
To determine whether bacterial contact would be sufficient to induce phagocyte cell death, phagocytosis was blocked by Cyto D (17). No reduction in the monocyte fraction (Fig. 1B, last column) was observed in either infected group, suggesting phagocytosis as an essential trigger of monocyte cell death.
To address the question of whether cell death would be less pronounced if monocytes were adhered and matured, we compared freshly isolated monocytes with cells that had been precultured for 48 h before bacterial challenge. In the latter group, we found slightly but not significantly diminished phagocytic activities (Table 1, lower) but no assimilation in cell death rates (Fig. 1C, last column) (p < 0.05). As previously described, heat-inactivated E. coli did not induce cell death in PBMO (23,24) or CBMO. The number of lymphocytes changed neither in PBMC nor CBMC 24 h after infection (data not shown).
Reduced E. coli-mediated apoptotic cell death in CBMO.
Four hours after phagocytosis, Annexin V binding was amplified from 254 ± 35 MFI to 341 ± 140 MFI (MOI 1:5) and 435 ± 173 MFI (MOI 1:50) on PBMO and from 156 ± 106 MFI to 187 ± 86 MFI (MOI 1:5) and 186 ± 90 MFI (MOI 1:50) on CBMO (p < 0.05 vs PBMO for MOI 1:50) (Fig. 2B). 24 h after phagocytosis, Annexin V binding was up-regulated almost threefold on PBMO while only marginally enhanced on CBMO (p < 0.05 vs PBMO) (Fig. 2C). Annexin V-positive monocytes were PI negative, indicating apoptosis rather than necrosis (data not shown). In the same experimental setup, hypodiploid DNA was quantified by way of PI stain (Fig. 3A and B), and nuclear fragmentation was detected by DAPI stain (Fig. 3C). DNA degradation was found in 56 ± 15% of PBMO versus 9 ± 6% in CBMO 24 h after phagocytosis (p < 0.05) (Fig. 3B); 50 ± 25% of PBMO versus 7 ± 3% CBMO showed a hypodiploid DNA content (MOI 1:5; p < 0.05) (Fig. 3B). Condensed nuclei as signs of nuclear fragmentation were only detectable in PBMO after phagocytosis (Fig. 3C).

Annexin V staining after bacterial infection. Monocytes were purified and incubated with GFP-labeled E. coli for 1 h. Extracellular bacteria were removed, monocytes were incubated for 4 (B) and 24 h (C), stained with Annexin V, and analyzed by FACS. (A) One of five representative experiments with 24 h incubation. PBMO, (▪); CBMO, (□), MOI 1:50; *p < 0.05 vs CBMO.

Intracellular DNA content and nuclear fragmentation after bacterial infection. (A) PBMΟ (left) and CBMΟ (right) analyzed for intracellular hypodiploid DNA content 24 h after challenge with stimuli indicated (MOI 1:50, staurosporine 1 μmol, mitomycin 50 μg/mL). (B) PBMO, (▪); CBMO, (□); n = 5, *p < 0.05 vs CBMO. (C) Nuclear fragmentation was detected by way of DAPI stain. Untreated PBMO (upper left), untreated CBMO (lower left), and CBMO exposed to E. coli (lower right) show normal chromatin pattern. PBMO exposed to E. coli show intense DAPI staining of condensed nuclei as a sign of apoptosis.
To address the question of whether CBMO would intrinsically be less sensitive toward apoptosis, we used staurosporine and mitomycin as common pro-apoptotic stimuli (25–27). Hypodiploid DNA fractions in monocytes were detectable at similar rates (p > 0.05) (Fig. 3B).
Diminished E. coli-mediated caspase-8 and -9 activation in CBMO.
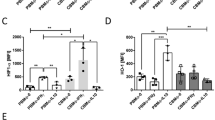
One h after phagocytosis, caspase-8 in PBMO was up-regulated from 14 ± 7 MFI (control group) to 37 ± 10 MFI (p < 0.05), whereas up-regulation on CBMO was marginal (NS vs control, p < 0.05 vs infected PBMO) (Fig. 4). Caspase-8 activation could not be blocked by the pan-caspase inhibitor zVAD-fmk (Fig. 4A).

Intracellular activation of caspase-8 and -9. Monocytes were purified and incubated with GFP-labeled E. coli for 1 h. Extracellular bacteria were removed, monocytes were incubated for a consecutive hour, stained with caspase-8 (A) or caspase-9 (B) specific fluorescence-labeled substrates, and analyzed. To block caspase activity, caspase inhibitor zVAD-fmk (50 μM) was used 1 h before infection (PBMO, ▪; CBMO, □; n = 5, *p < 0.05 vs CBMO, §p < 0.05 vs infected monocytes or vs zVAD preincubated monocytes).
After phagocytosis, activated caspase-9 in PBMO was up-regulated from 15 ± 7 MFI (control group) to 44 ± 15 MFI (p < 0.05), whereas its up-regulation in CBMO was diminished (p < 0.05 vs PBMO). In contrast with caspase-8, zVAD diminished E. coli-mediated caspase-9 activation (p < 0.05 vs infected PBMO) (Fig. 4B).
CD95L MRNA, Intracellular CD95L Content, and CD95 Expression on Monocytes.
Basal intracellular CD95L content in both monocyte populations was low (Fig. 5). Four hours after phagocytosis, CD95L in PBMO was up-regulated to 24 ± 2 MFI (p < 0.05 vs control) and 26 ± 7 MFI in CBMO (NS vs control). Twenty-four h after phagocytosis, intracellular CD95L content in PBMO nearly doubled (42 ± 10 MFI, p < 0.05 vs 4 h), which was in contrast with CBMO (31 ± 13 MFI, NS vs 4 h) (Fig. 5A). On the transcriptional level, we found a sevenfold increase of CD95L mRNA content in PBMO 4 h after phagocytosis, whereas no up-regulation in CBMO was detectable (p < 0.05 vs PBMO) (Fig. 5B). Monocyte CD95 receptor surface expression was not influenced by E. coli-mediated phagocytosis in the intervals tested (Fig. 5C).

CD95, CD95L, and CD95L mRNA expression. Monocytes were purified and incubated with GFP-labeled E. coli for 1 h. Extracellular bacteria were washed away, monocytes were incubated for the times indicated, and either stained with (A) anti-CD95 (PBMO, ▪; CBMO, □; n = 5, §p < 0.05 vs infected monocytes) or (C) anti-CD95. (B) mRNA-content was quantified with real-time PCR (n = 3, *p < 0.05 vs CBMO).
DISCUSSION
PICD of phagocytes is a common phenomenon upon interaction with bacteria, mainly induced by apoptosis (10). Diminished spontaneous apoptosis of neonatal neutrophils has already been described (13,14,28), which may contribute to persisting inflammation in preterm infants (13). In contrast, neonatal have not yet been compared with adult monocytes with regard to PICD.
Our results indicate that E. coli-mediated PICD is diminished in CBMO (Fig. 1). Characteristic features of apoptosis, that is, phosphatidylserine externalization (Fig. 2), DNA fragmentation (Fig. 3), capase-8- and -9 activation (Fig. 4), and CD95L up-regulation (Fig. 5), were found to be reduced in CBMO compared with PBMO. Our data suggest, however, that in contrast with neonatal neutrophils (13, 14, 28), neonatal monocytes are not generally more resistant toward apoptotic signals because staurosporine- and mitomycin-induced cell death was equivalent to cells from adults (Fig. 3). Both agents induce apoptosis by way of several pro-apoptotic mechanisms (25–27). Thus, it is likely that an alteration of more than one signaling pathway, involving cascades for apoptosis or survival, is responsible for reduced PICD in CBMO.
Apoptosis may be mediated by two main pathways: an extrinsic pathway, initiated by CD95 activation and mediated by caspase-8, and an intrinsic pathway, activated by various stimuli resulting in mitochondrial cytochrome c release and activation of caspase-9 (2). Both extrinsic (23) and intrinsic (24,29) pathways are considered to be relevant for PICD-mediated apoptosis induction. We showed that in CBMO, caspase-8 and -9 were activated to a weaker extent upon phagocytosis of E. coli and that this could be partially inhibited by the caspase inhibitor zVAD (Fig. 4).
Our data suggest that the CD95/CD95L pathway may be involved in reduced PICD in CBMO. Compared with CBMO, CD95L protein and mRNA were up-regulated more strongly in PBMO (Fig. 5). Whether this reduced CD95L up-regulation in CBMO is specific for E. coli or whether blockage of the CD95/CD95L pathway would be sufficient for modulating monocyte apoptosis in PBMO cannot be answered by our experiments and requires further investigation. Basal monocyte CD95/CD95L expression was equivalent (Fig. 5), whereas Drenou et al. (30) found lower expression in unseparated CBMC compared with adults. Intracellular ROS production, one important mediator of PICD through enhancement of caspase-8 activation (11), was found to be equivalent in CBMO compared with PBMO, confirming earlier results (22). Taken together, alteration of the extrinsic apoptosis pathway may be relevant for reduced PICD in CBMO.
After phagocytosis of Staphylococcus aureus, monocytes may release CD95L, which induces fratricidal apoptotic cell death in monocytes and bystander cells (i.e., neutrophils) (23). With regard to the substantially decreased CD95L content and diminished CD95L mRNA up-regulation in infected CBMO, apoptosis induction may not only be reduced in the phagocyting monocytes but also in bystander cells. Similar to our findings, monocytes expressed high CD95 levels, which did not change substantially after bacterial infection (Fig. 5C) (23).
With respect to PICD, we interpret our data with caution. We used an in vitro system with relatively small sample sizes and one bacterial species. Furthermore, for reasons of sample volumes required, we only have investigated cord blood and not peripheral blood of newborns after labor. Our model may therefore not properly reflect the neonatal situation several days after birth. We investigated a heterogeneous group of full-term neonates, and although none of them developed early onset infection, we cannot rule out effects of antenatal administration of antibiotics or steroids on the neonates' immunologic profile. To examine these parameters, as well as interindividual variations, further experiments elucidating pathophysiologic mechanisms are required.
Diminished PICD in CBMO fits into the broader picture of quantitative and qualitative differences between CBMO and PBMO. Neonatal monocytes express human leukocyte antigen-DR, toll-like receptor-2 (TLR2), and TLR4 in lower densities (31–35) and produce less interferon-gamma (IFN-gamma) and interleukin-12 (IL-12) (36). Moreover, they are less sensitive to IFN-gamma induced phenotypic and functional changes with respect to costimulatory signals by way of B7-1 (CD86) and B7-2 (CD80), resulting in diminished neonatal T-cell activation compared with adults (31,32). With regard to monocytes and monocyte-derived macrophages, a septic scenario in neonates versus adults would include diminished cell numbers with comparable phagocytic and ROS-producing activities (Tables 1 and 2) (15,22) but with deficiencies in costimulation, activation, and IFN-gamma production. Because neonatal monocytes are reduced in number compared with adults (Fig. 1B), their increased survival number in vivo may provide a physiologic counterbalance their impaired costimulatory potential.
A more severe parainfectious systemic inflammatory response is observed clinically in neonates than adults (37), and CBMO were found less capable to mediate inhibitory effects by way of hyporesponsiveness toward IL-10 (38). Together with findings of elevated pro-inflammatory cytokine levels in septic preterm infants (37), our observations suggest their capacity to terminate inflammatory reactions is also impaired. Less monocytic PICD in neonatal sepsis could contribute to secondary inflammatory cascades of previously activated and nondeleted monocytes, which results in prolonged inflammation and organ destruction, a phenomenon that is observed quite frequently in preterm infants (37,39). A recent study on adult septic patients suggests that an early increase in monocyte apoptosis may be associated with improved survival (8,9). These studies point toward the important role of monocytes in the orchestration of postphagocytic reactions. After elucidation of the mechanisms underlying reduced neonatal monocyte PICD, modifications of this feature may be a future target for sepsis intervention.
Abbreviations
- CBMC:
-
cord blood mononuclear cells
- CBMO:
-
cord blood monocytes
- Cyto D:
-
cytochalasin D
- DAPI:
-
4′,6-diamidino-2-phenylindole-dihydrochloride
- GFP:
-
green fluorescent protein
- MFI:
-
mean fluorescence intensity
- MOI:
-
multiplicity of infection
- PBMC:
-
peripheral blood mononuclear cells
- PBMO:
-
peripheral blood monocytes
- PI:
-
propidium iodide
- PICD:
-
phagocytosis-induced cell death
- ROS:
-
reactive oxygen species
References
Schrag SJ, Stoll BJ 2006 Early-onset neonatal sepsis in the era of widespread intrapartum chemoprophylaxis. Pediatr Infect Dis J 25: 939–940
Hotchkiss RS, Nicholson DW 2006 Apoptosis and caspases regulate death and inflammation in sepsis. Nat Rev Immunol 6: 813–822
Toti P, De Felice C, Occhini R, Schuerfeld K, Stumpo M, Epistolato MC, Vatti R, Buonocore G 2004 Spleen depletion in neonatal sepsis and chorioamnionitis. Am J Clin Pathol 122: 765–771
Doughty L, Clark RS, Kaplan SS, Sasser H, Carcillo J 2002 sFas and sFas ligand and pediatric sepsis-induced multiple organ failure syndrome. Pediatr Res 52: 922–927
Hotchkiss RS, Swanson PE, Freeman BD, Tinsley KW, Cobb JP, Matuschak GM, Buchman TG, Karl IE 1999 Apoptotic cell death in patients with sepsis, shock, and multiple organ dysfunction. Crit Care Med 27: 1230–1251
Hotchkiss RS, Tinsley KW, Swanson PE, Grayson MH, Osborne DF, Wagner TH, Cobb JP, Coopersmith C, Karl IE 2002 Depletion of dendritic cells, but not macrophages, in patients with sepsis. J Immunol 168: 2493–2500
Hotchkiss RS, Tinsley KW, Swanson PE, Schmieg RE Jr, Hui JJ, Chang KC, Osborne DF, Freeman BD, Cobb JP, Buchman TG, Karl IE 2001 Sepsis-induced apoptosis causes progressive profound depletion of B and CD4+ T lymphocytes in humans. J Immunol 166: 6952–6963
Giamarellos-Bourboulis EJ, Routsi C, Plachouras D, Markaki V, Raftogiannis M, Zervakis D, Koussoulas V, Orfanos S, Kotanidou A, Armaganidis A, Roussos C, Giamarellou H 2006 Early apoptosis of blood monocytes in the septic host: is it a mechanism of protection in the event of septic shock?. Crit Care 10: R76
Moraes TJ, Downey GP 2006 Death of the septic monocyte: is more better?. Crit Care 10: 146
DeLeo FR 2004 Modulation of phagocyte apoptosis by bacterial pathogens. Apoptosis 9: 399–413
Zhang B, Hirahashi J, Cullere X, Mayadas TN 2003 Elucidation of molecular events leading to neutrophil apoptosis following phagocytosis. J Biol Chem 278: 28443–28454
Skrzeczynska J, Kobylarz K, Hartwich Z, Zembala M, Pryjma J 2002 CD14+ CD16+ monocytes in the course of sepsis in neonates and small children: monitoring and functional studies. Scand J Immunol 55: 629–638
Hanna N, Vasquez P, Pham P, Heck DE, Laskin JD, Laskin DL, Weinberger B 2005 Mechanisms underlying reduced apoptosis in neonatal neutrophils. Pediatr Res 57: 56–62
Koenig JM, Stegner JJ, Schmeck AC, Saxonhouse MA, Kenigsberg LE 2005 Neonatal neutrophils with prolonged survival exhibit enhanced inflammatory and cytotoxic responsiveness. Pediatr Res 57: 424–429
Gille C, Spring B, Tewes L, Poets CF, Orlikowsky T 2006 A new method to quantify phagocytosis and intracellular degradation using green fluorescent protein-labeled Escherichia coli: comparison of cord blood macrophages and peripheral blood macrophages of healthy adults. Cytometry A 69: 152–154
Dehio M, Knorre A, Lanz C, Dehio C 1998 Construction of versatile high-level expression vectors for Bartonella henselae and the use of green fluorescent protein as a new expression marker. Gene 215: 223–229
Gille C, Spring B, Bernhard W, Gebhard C, Basile D, Lauber K, Poets CF, Orlikowsky TW 2007 Differential effect of surfactant and its saturated phosphatidylcholines on human blood macrophages. J Lipid Res 48: 307–317
Gerber CE, Kuci S, Zipfel M, Niethammer D, Bruchelt G 1996 Phagocytic activity and oxidative burst of granulocytes in persons with myeloperoxidase deficiency. Eur J Clin Chem Clin Biochem 34: 901–908
Brown SB, Savill J 1999 Phagocytosis triggers macrophage release of Fas ligand and induces apoptosis of bystander leukocytes. J Immunol 162: 480–485
Nicoletti I, Migliorati G, Pagliacci MC, Grignani F, Riccardi C 1991 A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J Immunol Methods 139: 271–279
Loeffler J, Bauer R, Hebart H, Douek DC, Rauser G, Bader P, Einsele H 2002 Quantification of T-cell receptor excision circle DNA using fluorescence resonance energy transfer and the LightCycler system. J Immunol Methods 271: 167–175
Speer CP, Ambruso DR, Grimsley J, Johnston RB Jr 1985 Oxidative metabolism in cord blood monocytes and monocyte-derived macrophages. Infect Immun 50: 919–921
Baran J, Weglarczyk K, Mysiak M, Guzik K, Ernst M, Flad HD, Pryjma J 2001 Fas (CD95)-Fas ligand interactions are responsible for monocyte apoptosis occurring as a result of phagocytosis and killing of Staphylococcus aureus. Infect Immun 69: 1287–1297
Hacker H, Furmann C, Wagner H, Hacker G 2002 Caspase-9/-3 activation and apoptosis are induced in mouse macrophages upon ingestion and digestion of Escherichia coli bacteria. J Immunol 169: 3172–3179
Yamaki K, Hong J, Hiraizumi K, Ahn JW, Zee O, Ohuchi K 2002 Participation of various kinases in staurosporine induced apoptosis of RAW 264.7 cells. J Pharm Pharmacol 54: 1535–1544
Pirnia F, Schneider E, Betticher DC, Borner MM 2002 Mitomycin C induces apoptosis and caspase-8 and -9 processing through a caspase-3 and Fas-independent pathway. Cell Death Differ 9: 905–914
Kang YH, Lee KA, Ryu CJ, Lee HG, Lim JS, Park SN, Paik SG, Yoon DY 2006 Mitomycin C induces apoptosis via Fas/FasL dependent pathway and suppression of IL-18 in cervical carcinoma cells. Cancer Lett 237: 33–44
Allgaier B, Shi M, Luo D, Koenig JM 1998 Spontaneous and Fas-mediated apoptosis are diminished in umbilical cord blood neutrophils compared with adult neutrophils. J Leukoc Biol 64: 331–336
Weglarczyk K, Baran J, Zembala M, Pryjma J 2004 Caspase-8 activation precedes alterations of mitochondrial membrane potential during monocyte apoptosis induced by phagocytosis and killing of Staphylococcus aureus. Infect Immun 72: 2590–2597
Drenou B, Choqueux C, El Ghalbzouri A, Blancheteau V, Toubert A, Charron D, Mooney N 1998 Characterisation of the roles of CD95 and CD95 ligand in cord blood. Bone Marrow Transplant 22: S44–S47
Orlikowsky TW, Spring B, Dannecker GE, Niethammer D, Poets CF, Hoffmann MK 2003 Expression and regulation of B7 family molecules on macrophages (MPhi) in preterm and term neonatal cord blood and peripheral blood of adults. Cytometry B Clin Cytom 53: 40–47
Orlikowsky TW, Dannecker GE, Spring B, Eichner M, Hoffmann MK, Poets CF 2005 Effect of dexamethasone on B7 regulation and T cell activation in neonates and adults. Pediatr Res 57: 656–661
Forster-Waldl E, Sadeghi K, Tamandl D, Gerhold B, Hallwirth U, Rohrmeister K, Hayde M, Prusa AR, Herkner K, Boltz-Nitulescu G, Pollak A, Spittler A 2005 Monocyte toll-like receptor 4 expression and LPS-induced cytokine production increase during gestational aging. Pediatr Res 58: 121–124
Viemann D, Dubbel G, Schleifenbaum S, Harms E, Sorg C, Roth J 2005 Expression of toll-like receptors in neonatal sepsis. Pediatr Res 58: 654–659
Sadeghi K, Berger A, Langgartner M, Prusa AR, Hayde M, Herkner K, Pollak A, Spittler A, Forster-Waldl E 2007 Immaturity of infection control in preterm and term newborns is associated with impaired toll-like receptor signaling. J Infect Dis 195: 296–302
Taylor S, Bryson YJ 1985 Impaired production of gamma-interferon by newborn cells in vitro is due to a functionally immature macrophage. J Immunol 134: 1493–1497
Schultz C, Rott C, Temming P, Schlenke P, Moller JC, Bucsky P 2002 Enhanced interleukin-6 and interleukin-8 synthesis in term and preterm infants. Pediatr Res 51: 317–322
Gille C, Spring B, Tewes LJ, Loffler J, Dannecker GE, Hoffmann MK, Eichner M, Poets CF, Orlikowsky TW 2006 Diminished response to interleukin-10 and reduced antibody-dependent cellular cytotoxicity of cord blood monocyte-derived macrophages. Pediatr Res 60: 152–157
Dammann O, Leviton A 2006 Inflammation, brain damage and visual dysfunction in preterm infants. Semin Fetal Neonatal Med 11: 363–368
Acknowledgements
Thanks to Harald Abele, M.D., and Regina Harms, Department of Obstetrics and Gynaecology, for coordinating the cord blood sampling. Thanks to Andrea Schäfer and Hanna Hartmann, Ph.D., Department of Microbiology, for technical help, Michael Urschitz, M.D., Department of Neonatology, for help with statistics, and Christopher Pynn, Ph.D., Department of Neonatology for critical reading of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
This work was supported by the Deutsche Sepsisgesellschaft and Sanitätsrat Dr. Alexander Hübner und Gemahlin Stiftung (Deutscher Stifterverband).
Rights and permissions
About this article
Cite this article
Gille, C., Leiber, A., Spring, B. et al. Diminished Phagocytosis-Induced Cell Death (PICD) in Neonatal Monocytes upon Infection with Escherichia coli. Pediatr Res 63, 33–38 (2008). https://doi.org/10.1203/PDR.0b013e31815b8e9f
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/PDR.0b013e31815b8e9f
This article is cited by
-
Impact of perinatal administration of probiotics on immune cell composition in neonatal mice
Pediatric Research (2024)
-
Impaired cellular energy metabolism in cord blood macrophages contributes to abortive response toward inflammatory threats
Nature Communications (2019)
-
Cord blood granulocytic myeloid-derived suppressor cells impair monocyte T cell stimulatory capacity and response to bacterial stimulation
Pediatric Research (2019)
-
Neonatal monocytes express antiapoptotic pattern of Bcl-2 proteins and show diminished apoptosis upon infection with Escherichia coli
Pediatric Research (2014)
-
The CD95/CD95L pathway is involved in phagocytosis-induced cell death of monocytes and may account for sustained inflammation in neonates
Pediatric Research (2013)
