
Abstract
Prenatal nicotine exposure influences neuronal development including effects on several neurotransmitter systems. It also attenuates the ventilatory response to hypoxia, known to require a functional substance P-ergic system. Previous studies have shown that nicotine increases the risk for sudden infant death syndrome (SIDS) by 4-fold, and that SIDS-victims have elevated brainstem levels of substance P. We, therefore, studied the effect of prenatal nicotine exposure on the levels of substance P-like immunoreactivity by RIA in the brain in newborn rat pups. The expression of the substance P precursor preprotachykinin A mRNA was also determined by real-time reverse transcriptase-polymerase chain reaction in carotid body, in petrosal/jugular and trigeminal ganglia, in cervical and lumbar dorsal root ganglia, and in the brainstem. We found that prenatal nicotine exposure increased levels of substance P-like immunoreactivity in the brainstem without changing levels in other parts of the brain or in the adrenals. Furthermore, mRNA levels were increased in the carotid bodies and in the petrosal ganglia, in contrast to the decreased levels in the cervical dorsal root ganglia. We conclude that nicotine causes alterations in the substance P-ergic system in the brainstem, possibly linked to the increased risk for SIDS after prenatal nicotine exposure.
Similar content being viewed by others

Main
Nicotine exposure increases the risk of sudden infant death syndrome (SIDS) by up to 4-fold, thereby being the strongest behavioral risk for SIDS. Although tobacco smoke contains a wide variety of potentially fetotoxic substances, and although smoking has numerous covariables that can contribute to adverse outcomes, animal studies have pointed to nicotine itself as a neuroteratogen, producing an abnormal CNS development (1,2). From a respiratory point of view, studies in newborn rat and mouse of the effects of prenatal nicotine exposure have shown a deficient ventilatory response to hypoxia (3,4) and an impaired autoresuscitation in newborn rat during anoxia (5).
Prenatal nicotine has been demonstrated to disrupt several neurotransmitter systems (3,6,7), but the tachykinin substance P has not been investigated in this aspect. However, substance P may be of potential interest because it is known to be involved in the central control of respiration in the newborn mammal. For instance, central administration of substance P increases ventilation in rabbit pups (8), and in vitro experiments in the newborn rat show a stimulatory effect on respiration by substance P (9,10). In addition, our group has shown that newborn transgenic mice, lacking substance P and neurokinin A, display an abnormal hypoxic response and an impaired plasticity of the central respiratory network (11). Similarly, blocking the functional substance P-ergic transmission via its preferred receptor, the neurokinin-1 receptor (NK-1), affects the hypoxic ventilatory response (12). Accordingly, NK-1 receptors are present in respiration-related ganglia and in their corresponding areas in the brainstem, and they are also known to be involved in the control of rhythmicity of respiration (for review, see Ref. 13). Furthermore, substance P is considered to be of relatively greater importance in the newborn period (8,14–17), and mRNA encoding the substance P-precursor, preprotachykinin A (PPT-A), increases in the nucleus tractus solitarii (NTS) immediately after birth in rabbits (18) and rats (19). Unlike the abundant expression e.g., in the brainstem, PPT-A mRNA expression has not been detected in the carotid bodies of adult mice (20). The carotid body contains glomus cells or type I cells known to be involved in chemoreception and is innervated by the carotid sinus nerve with afferent projections via the petrosal and jugular ganglia to respiratory related areas in the brainstem (21,22). Nicotine, through nicotinic acetylcholinergic receptors (nAChrs), also modulates the hypoxic chemosensitivity in the rat carotid body (3,23).
Nicotine, and exposure to cigarette smoke, has been shown to decrease substance P-like immunoreactivity (P-LI) in the brain of adult rat (24), and substance P is considered to be of major importance for the action of nicotine on the nervous system (25,26). In view of these facts, we hypothesized that prenatal exposure to nicotine may cause alterations in this neurotransmitter system. To test this, we determined substance P-LI in the brain and adrenals as well as the PPT-A mRNA expression in brainstem, carotid bodies, sensory ganglia involved in respiratory control and in dorsal root ganglia (DRGs) unrelated to respiration, in newborn rat pups prenatally exposed to nicotine.
MATERIALS AND METHODS
Animals.
Sprague-Dawley rat pups of both sexes (B&K Universal, Sollentuna, Sweden) were studied on postnatal d 1 (P1). All pups were born naturally and left with their mothers until assigned age. Animals were kept on a regular 12 h light:12 h dark cycle and treated according to the guidelines approved by the local ethics committee (see below). The number of pups studied in each group (nicotine and sodium chloride) was n = 8 being from two litters for the RIA (27), and n = 12 for each group (nicotine and sodium chloride) for the real-time reverse transcriptase-polymerase chain reaction (RT-PCR) analysis being from three litters.
Administration of nicotine.
Osmotic minipumps (Model 2ML4, Alzet, Palo Alto, CA) were used to administer nicotine to the fetuses. Insertion of the pump was made on the 5th d of gestation after lightly anesthetizing the dam with ether. The osmotic pump was placed s.c. on the back, the incision was closed with a suture, and the animals were returned to their cages for recovery. The pumps contained either nicotine bitartrate (Sigma Chemical Co., St. Louis, MO) dissolved in sodium chloride, or only sodium chloride for the control group. The concentration of nicotine was designed to deliver 3 mg nicotine free base/kg bodyweight per day, calculated for the initial weight of the dam and thus decreasing toward the end of gestation. Based on previous studies, a daily dose of 2 or 6 mg/kg is considered to produce effects comparable to moderate or heavy smoking, respectively, in man (25,28,29). The pumps deliver their content for approximately 17 d (information provided by the manufacturer), thus lasting the entire remaining gestational period and postnatally until the time studied. Osmotic minipumps offer a safe route of administration and, unlike injected nicotine, do not cause any overt signs of hypoxia or ischemia, such as blanching of the skin or cyanosis, nor are there any long term changes in maternal blood pressure or heart rate (30).
RIA.
Substance P-LI was determined by RIA, as previously described in detail (31). The detection limit of the substance P-assay was 10 fmol per mL of sample (sample volume 0.1 mL). Tissue concentrations of substance P are expressed in pmol of substance P per g of tissue wet weight. Animals used for RIA were killed by decapitation and brains were rapidly dissected out. They were thereafter divided into the hindbrain including the cerebellum (referred to as hindbrain) and structures cranial thereof (referred to as forebrain). Sections were made by hand using the following landmarks: the hindbrain was caudally limited by the sulcus located at the cranial end of the pyramidal decussation. Rostrally, the hindbrain was cut between the primary sulcus and the posterior lobe of the pituitary, the cerebellum was not removed. The tissues samples were weighed and frozen on dry ice immediately after dissection. Extraction was performed at 100°C in distilled water and thereafter in 1.0 M acetic acid (32,33). The neutral and acid extracts of each sample were pooled, lyophilized, and analyzed by RIA for substance P-LI using antiserum SP2 (34,35).
Real-time RT-PCR.
The following tissues were collected and immediately frozen on dry ice; carotid bodies, petrosal together with jugular ganglia, trigeminal ganglia, and cervical and lumbar DRGs. The carotid bodies, the petrosal and trigeminal ganglia contain substance P and project to NTS and other brainstem areas (22,36). The lumbar DRGs were included to monitor PPT-A mRNA levels in sensory ganglia definitely not involved in respiratory control. The cervical DRGs served as ganglia with projections to the lower brainstem but similarly without direct involvement in respiratory control (37). Also, total RNA was isolated from the brainstem at three different levels using transversal cryostat sections. The first level was at the rostral end of the lateral reticular nucleus, the second level at the locus coeruleus, and the third level at NTS. Tissue homogenisation and RNA extraction (RNAas Mini kit, Qiagen, Stanford, CA) were both performed according to the protocol supplied by the manufacturer. Relative quantification of PPT-A gene expression was performed by real-time RT-PCR (iQ5 Real-Time Detection System, Bio-Rad Laboratories, Sundbyberg, Sweden) by using iScript 1-step RT-PCR SYBR (Bio-Rad Laboratories, Sundbyberg, Sweden). Primer pairs for the PPT-A gene were PPT-A 1 upstream 5′-ATT ATT GGT CCG ACT GGT CCG-3′ and downstream 5′-CCC GCT TGC CCA TTA-3′. A commercial kit, Quantum RNA classical 18S internal standard (Ambion, Austin, TX), was used for 18S determination for standardization of the amount of PPT-A mRNA in the different samples. PCR protocol: cycle 1, (×1) 50°C for 10:00. cycle 2, (×1) 95°C for 5:00. cycle 3, (×45). step 1, 95°C for 00:10 and step 2, 57°C for 00:30. cycle 4, (×1) 95°C for 1:00. cycle 5, (×1) 55°C for 1:00. cycle 6, (×80) 55°C for 00:10 (increased setpoint temperature after cycle 2 by 0.5°C). All the reactions were performed in triplicate in a total volume of 50 μL. Relative quantification of gene expression was performed by using the delta Ct method. The Ct values (i.e., the point at which the fluorescence crosses the threshold) were converted into arbitrary quantities of the initial template of a given sample.
Statistical considerations.
All statistical analyses were made on absolute values before normalization for graphical clarity. For RIA and real-time RT-PCR, we used one-way ANOVA (Statistica software, StatSoft Scandinavia AB, Uppsala, Sweden), where ANOVA indicated significant interactions, a planned comparisons test was performed with a confidence level of p < 0.05 considered as significant.
Ethical considerations.
These experiments have been approved by the local ethics committee, i.e., Stockholms norra djurförsöksetiska nämnd (Dnr N321/98 and N40/04).
RESULTS
Litter sizes and birth-weight did not significantly vary between nicotine-exposed and control pups (data not shown). Similarly, there were no overt differences in maternal care taking of the dams or behavior of rat pups. No differences were seen between male and female pups for any parameter.
Substance P levels.
The levels of substance P-LI in the brainstem and the forebrain were determined using RIA. After prenatal nicotine exposure, the levels of substance P-LI in the hindbrain increased from 17.1 ± 4.0 in control animals to 43.3 ± 14.9 pmol/g (p = 0.007, Fig. 1A). In contrast, substance P-LI in forebrain did not differ between groups (10.5 ± 5.6 and 10.8 ± 4.4 pmol/g, respectively, Fig. 1B). Nor was a significant difference observed in the adrenals (1.13 ± 0.85 and 0.60 ± 0.47 pmol/g, respectively), although there was a tendency toward increased substance P-LI in the nicotine-treated animals (Fig. 1C).

SP-LI in hindbrain (A), cortex (B), and adrenals (C) in control and nicotine-treated animals (3 mg/kg × d). The box includes the 25th to 75th percentiles with the median value indicated as a line within the box. The whiskers above and below the box indicate the 10th and 90th percentile. A statistically significant difference was seen only in the hindbrain (*p =0.007).
PPT-A mRNA levels.
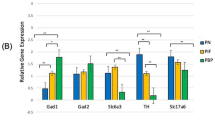
PPT-A mRNA was detected in the carotid bodies of newborn rat pups, and prenatal nicotine exposure markedly increased this expression (p = 0.003, Fig. 2A). An even larger increase was seen in the petrosal/jugular ganglia (p < 0.001, Fig. 2B). In contrast, PPT-A mRNA levels were significantly decreased in the cervical DRGs of nicotine-exposed pups (p = 0.003, Fig. 2C), and a tendency toward a decrease was also seen in the trigeminal ganglia (Fig. 2D). There were no differences between experimental groups in the brainstem at any level (locus coeruleus, NTS or lateral reticular nucleus) or in the lumbar DRGs (Fig. 2E and F, respectively).

A–F, PPT-A mRNA expression increased after nicotine exposure in the carotid bodies and petrosal ganglia [*p = 0.003 (A) and **p < 0.001 (B), respectively]. The cervical DRGs [†p = 0.003, (C)] and the trigeminal ganglia (D), both involved in sensory input, displayed an opposite reaction to nicotine with decreased levels of PPT-A mRNA (significant only in the cervical DRGs). No differences were seen between experimental groups in the lumbar DRGs (E) or in the brainstem [(the lateral reticular nucleus, the locus coeruleus, and NTS are displayed pooled together, (F)]. The box includes the 25–75th percentiles with the median value indicated as a line within the box. The whiskers above and below the box indicate the 10th and 90th percentile. Please note different scaling. Y-axis denotes Ct values converted into arbitrary quantities of initial template of a given sample.
DISCUSSION
In this study, we demonstrate elevated substance P-LI in the brainstem and increased PPT-A mRNA expression in the peripheral arterial chemoreceptors in newborn rat after prenatal nicotine exposure, in fact a 5-fold increase for the carotid body and a 40-fold increase in the petrosal/jugular ganglia. Nicotinic effects on the substance P-ergic system in the adult rat brain have been previously described with decreased substance P-LI in discrete brain regions after both acute and chronic exposure to nicotine (24,38). This may represent a regional differentiation of brain nuclei or a developmental difference.
The increased levels of substance P-LI in the brainstem could indicate a decreased release and a subsequent accumulation of substance P in presynaptic afferent nerve terminals. This may be accomplished by a disruption of feedback loops of acetylcholinergic neurons, known to trigger substance P release in the brain (24) and gut (39). Furthermore, it has been shown that perinatal nicotine exposure causes a blunted peripheral arterial chemosensitivity in P3 rat pups in the presence of a normally functioning carotid sinus nerve, indicating a dynamic dysfunction at the brainstem level (40). However, the markedly increased PPT-A mRNA levels in carotid bodies and petrosal/jugular ganglia after nicotine treatment indicate that the effect on the substance P-ergic neurotransmission in respiration-related pathways is primarily not a decreased peptide release. As elevated transcript levels rather reflect a compensatory increase of synthesis after peptide release (41), the increased mRNA levels in the petrosal/jugular ganglia indicate an increased substance P release in the NTS. This may be a compensatory mechanism due to a perturbed postsynaptic function. Previous studies have also demonstrated an attenuated respiratory hypoxic response after blocking the substance P-ergic NK-1 receptors in the brainstem (12).
It is important to note that the brainstem tissue analyzed here with RIA does not only contain the NTS, with afferent input from the petrosal/jugular ganglia, but also the spinal trigeminal nucleus and other sensory subnuclei (37,42) all rich in substance P (43–45). Furthermore, the NTS has substance P inputs from other sources, e.g., the nodose ganglion (46). Thus, one possible explanation is that the petrosal/jugular input is quantitatively small and that the major effect of nicotine on substance P levels measured by RIA is on these other systems. In fact, we observed a markedly decreased (cervical DRGs), or a distinct tendency to decreased (trigeminal ganglia), PPT-A mRNA levels in the ganglia giving rise to most substance P in the lower brain stem. This suggests a differential effect of maternal nicotine on sensory neurons in general versus the respiratory carotid bodies–petrosal/jugular ganglia pathway, leading to regional differences within the brainstem. Such differences may exist also within specific nuclei, as demonstrated previously in the spinal cord (43,47).
Previous studies in adult rat have detected PPT-A mRNA and NK-1 receptors in the petrosal ganglia but not in the carotid bodies, even after prolonged hypoxia (20). We found PPT-A mRNA expression also in the carotid bodies of control animals at P1, indicating the importance of tachykinins during early development. The increased mRNA levels suggest increased substance P release, which could activate the petrosal neurons via presynaptic NK-1 receptors (20). These findings are in line with other studies showing different effects of nicotine depending on site of action (48,49) in relation to the heterogeneous distribution of nicotinic receptor subunits (50). Similarly, we could not detect any alterations in PPT-A mRNA expression between experimental groups in the brainstem at any level. These levels include at least three areas rich of substance P interneurons i.e., NTS, the periventricular central gray, and the marginal layers (43). Further studies are required to determine the effects on a cellular level.
The close connection between the acetylcholinergic neurotransmitter system and substance P neurotransmission has been demonstrated in a number of studies. Substance P is known to modulate nAChrs in the brain (51) through direct receptor interaction, but without directly binding to the agonist site of the receptor (52). The reverse situation, where presynaptic nAChrs mediate the release of substance P, has been demonstrated in myenteric neurons of the gut (39). Lower levels of muscarinic M2 receptors have also been described in infants who had succumbed to SIDS (53). Incidentally, these children have also been demonstrated to have increased concentrations of substance P in the medulla (14,54). Thus, it is possible that prenatal exposure to nicotine causes alterations in substance P-ergic neurotransmission via an acetylcholinergic pathway, and that these alterations impair the function of the respiratory network.
Taken together, our results offer the first description of increased levels of substance P-LI in the brainstem and alterations in PPT-A mRNA expression in structures related to respiration of newborn rat pups after prenatal exposure to nicotine. The present data do not support a decreased substance P release in respiratory nuclei in the brainstem, but they rather imply a perturbed postsynaptic function and a disruption of the respiration-related carotid bodies-petrosal ganglia pathway. This may in turn cause a deficient regulation of respiration in the newborn mammal and thereby contribute to the detrimental effects seen after intrauterine nicotine exposure.
Abbreviations
- DRGs:
-
dorsal root ganglia
- NK-1:
-
neurokinin 1-receptor
- NTS:
-
nucleus tractus solitarii
- RT-PCR:
-
reverse transcriptase-polymerase chain reaction
- PPT-A:
-
preprotachykinin A
References
Navarro HA, Seidler FJ, Eylers JP, Baker FE, Dobbins SS, Lappi SE, Slotkin TA 1989 Effects of prenatal nicotine exposure on development of central and peripheral cholinergic neurotransmitter systems. Evidence for cholinergic trophic influences in developing brain. J Pharmacol Exp Ther 251: 894–900
Wickström R 2007 Effects of nicotine during pregnancy: human and experimental evidence. Curr Neuropharmacol 5: 213–222
Holgert H, Hokfelt T, Hertzberg T, Lagercrantz H 1995 Functional and developmental studies of the peripheral arterial chemoreceptors in rat: effects of nicotine and possible relation to sudden infant death syndrome. Proc Natl Acad Sci U S A 92: 7575–7579
Slotkin TA, Lappi SE, McCook EC, Lorber BA, Seidler FJ 1995 Loss of neonatal hypoxia tolerance after prenatal nicotine exposure: implications for sudden infant death syndrome. Brain Res Bull 38: 69–75
Fewell JE, Smith FG 1998 Perinatal nicotine exposure impairs ability of newborn rats to autoresuscitate from apnea during hypoxia. J Appl Physiol 85: 2066–2074
Gauda EB, Cooper R, Akins PK, Wu G 2001 Prenatal nicotine affects catecholamine gene expression in newborn rat carotid body and petrosal ganglion. J Appl Physiol 91: 2157–2165
Dani JA, Heinemann S 1996 Molecular and cellular aspects of nicotine abuse. Neuron 16: 905–908
Yamamoto Y, Lagercrantz H 1985 Some effects of substance P on central respiratory control in rabbit pups. Acta Physiol Scand 124: 449–455
Monteau R, Ptak K, Broquere N, Hilaire G 1996 Tachykinins and central respiratory activity: an in vitro study on the newborn rat. Eur J Pharmacol 314: 41–50
Yamamoto Y, Onimaru H, Homma I 1992 Effect of substance P on respiratory rhythm and pre-inspiratory neurons in the ventrolateral structure of rostral medulla oblongata: an in vitro study. Brain Res 599: 272–276
Berner J, Shvarev Y, Lagercrantz H, Bilkei-Gorzo A, Hokfelt T, Wickstrom R 2007 Altered respiratory pattern and hypoxic response in transgenic newborn mice lacking the tachykinin-1 gene. J Appl Physiol 103: 552–559
Wickstrom HR, Berner J, Holgert H, Hokfelt T, Lagercrantz H 2004 Hypoxic response in newborn rat is attenuated by neurokinin-1 receptor blockade. Respir Physiol Neurobiol 140: 19–31
Feldman JL, Mitchell GS, Nattie EE 2003 Breathing: rhythmicity, plasticity, chemosensitivity. Annu Rev Neurosci 26: 239–266
Bergstrom L, Lagercrantz H, Terenius L 1984 Post-mortem analyses of neuropeptides in brains from sudden infant death victims. Brain Res 323: 279–285
Ptak K, Di Pasquale E, Monteau R 1999 Substance P and central respiratory activity: a comparative in vitro study on foetal and newborn rat. Brain Res Dev Brain Res 114: 217–227
Quirion R, Dam TV 1986 Ontogeny of substance P receptor binding sites in rat brain. J Neurosci 6: 2187–2199
Viemari JC, Burnet H, Bevengut M, Hilaire G 2003 Perinatal maturation of the mouse respiratory rhythm-generator: in vivo and in vitro studies. Eur J Neurosci 17: 1233–1244
Srinivasan M, Goiny M, Pantaleo T, Lagercrantz H, Brodin E, Runold M, Yamamoto Y 1991 Enhanced in vivo release of substance P in the nucleus tractus solitarii during hypoxia in the rabbit: role of peripheral input. Brain Res 546: 211–216
Wickstrom HR, Holgert H, Hokfelt T, Lagercrantz H 1999 Birth-related expression of c-fos, c-jun and substance P mRNAs in the rat brainstem and pia mater: possible relationship to changes in central chemosensitivity. Brain Res Dev Brain Res 112: 255–266
Gauda EB, Bamford OS, Northington FJ 1998 Lack of induction of substance P gene expression by hypoxia and absence of neurokinin 1-receptor mRNAs in the rat carotid body. J Auton Nerv Syst 74: 100–108
Ichikawa H 2002 Innervation of the carotid body: immunohistochemical, denervation, and retrograde tracing studies. Microsc Res Tech 59: 188–195
Finley JC, Katz DM 1992 The central organization of carotid body afferent projections to the brainstem of the rat. Brain Res 572: 108–116
Fidone SJ, Gonzalez C, Dinger BG, Hanson GR 1988 Mechanisms of chemotransmission in the mammalian carotid body. Prog Brain Res 74: 169–179
Naftchi NE, Maker H, Lapin E, Sleis J, Lajtha A, Leeman S 1988 Acute reduction of brain substance P induced by nicotine. Neurochem Res 13: 305–309
Lichtensteiger W, Ribary U, Schlumpf M, Odermatt B, Widmer HR 1988 Prenatal adverse effects of nicotine on the developing brain. Prog Brain Res 73: 137–157
Nasrat HA, Al-Hachim GM, Mahmood FA 1986 Perinatal effects of nicotine. Biol Neonate 49: 8–14
Lacoste B, Riad M, Descarries L 2006 Immunocytochemical evidence for the existence of substance P receptor (NK1) in serotonin neurons of rat and mouse dorsal raphe nucleus. Eur J Neurosci 23: 2947–2958
Murrin LC, Ferrer JR, Zeng WY, Haley NJ 1987 Nicotine administration to rats: methodological considerations. Life Sci 40: 1699–1708
Slotkin TA, Orband-Miller L, Queen KL, Whitmore WL, Seidler FJ 1987 Effects of prenatal nicotine exposure on biochemical development of rat brain regions: maternal drug infusions via osmotic minipumps. J Pharmacol Exp Ther 240: 602–611
Slotkin TA, McCook EC, Seidler FJ 1997 Cryptic brain cell injury caused by fetal nicotine exposure is associated with persistent elevations of c-fos protooncogene expression. Brain Res 750: 180–188
Brodin E, Rosen A, Theodorsson E, Jonczyk A, Sandberg BE, Brodin K 1994 Multiple molecular forms of tachykinins in rat spinal cord: a study comparing different extraction methods. Regul Pept 52: 97–110
Brodin E, Lindefors N, Dalsgaard CJ, Theodorsson-Norheim E, Rosell S 1986 Tachykinin multiplicity in rat central nervous system as studied using antisera raised against substance P and neurokinin A. Regul Pept 13: 253–272
Brodin E, Linderoth B, Gazelius B, Ungerstedt U 1987 In vivo release of substance P in cat dorsal horn studied with microdialysis. Neurosci Lett 76: 357–362
Thor KB, Helke CJ 1987 Serotonin- and substance P-containing projections to the nucleus tractus solitarii of the rat. J Comp Neurol 265: 275–293
Thor KB, Helke CJ 1989 Serotonin and substance P colocalization in medullary projections to the nucleus tractus solitarius: dual-colour immunohistochemistry combined with retrograde tracing. J Chem Neuroanat 2: 139–148
Zhang WB, Li JS, Li HM 1991 SP-like immunoreactivity in the primary trigeminal neurons projecting to the nucleus tractus solitarii. Brain Res 558: 87–89
Pfaller K, Arvidsson J 1988 Central distribution of trigeminal and upper cervical primary afferents in the rat studied by anterograde transport of horseradish peroxidase conjugated to wheat germ agglutinin. J Comp Neurol 268: 91–108
Fuxe K, Siegel RA, Andersson K, Eneroth P, Mascagni F, Agnati LF 1985 Acute continuous exposure to cigarette smoke produces discrete changes in cholecystokinin and substance P levels in the hypothalamus and preoptic area of the male rat. Acta Physiol Scand 125: 437–443
Schneider DA, Galligan JJ 2000 Presynaptic nicotinic acetylcholine receptors in the myenteric plexus of guinea pig intestine. Am J Physiol Gastrointest Liver Physiol 279: G528–G535
Bamford OS, Carroll JL 1999 Dynamic ventilatory responses in rats: normal development and effects of prenatal nicotine exposure. Respir Physiol 117: 29–40
Schalling M, Franco-Cereceda A, Hokfelt T, Persson H, Lundberg JM 1988 Increased neuropeptide Y messenger RNA and peptide in sympathetic ganglia after reserpine pretreatment. Eur J Pharmacol 156: 419–420
Kalia M 1981 Neurohistochemical methods in tracing central respiratory mechanisms. Fed Proc 40: 2365–2371
Ljungdahl A, Hokfelt T, Nilsson G 1978 Distribution of substance P-like immunoreactivity in the central nervous system of the rat—I. Cell bodies and nerve terminals. Neuroscience 3: 861–943
Kalia M, Fuxe K, Hokfelt T, Johansson O, Lang R, Ganten D, Cuello C, Terenius L 1984 Distribution of neuropeptide immunoreactive nerve terminals within the subnuclei of the nucleus of the tractus solitarius of the rat. J Comp Neurol 222: 409–444
Cuello AC, Del Fiacco M, Paxinos G 1978 The central and peripheral ends of the substance P-containing sensory neurones in the rat trigeminal system. Brain Res 152: 499–500
Helke CJ, O'Donohue TL, Jacobowitz DM 1980 Substance P as a baro- and chemoreceptor afferent neurotransmitter: immunocytochemical and neurochemical evidence in the rat. Peptides 1: 1–9
Hokfelt T, Ljungdahl A, Steinbusch H, Verhofstad A, Nilsson G, Brodin E, Pernow B, Goldstein M 1978 Immunohistochemical evidence of substance P-like immunoreactivity in some 5-hydroxytryptamine-containing neurons in the rat central nervous system. Neuroscience 3: 517–538
Damaj MI, Meyer EM, Martin BR 2000 The antinociceptive effects of alpha7 nicotinic agonists in an acute pain model. Neuropharmacology 39: 2785–2791
Molinero MT, Del Rio J 1987 Substance P, nicotinic acetylcholine receptors and antinociception in the rat. Neuropharmacology 26: 1715–1720
Albuquerque EX, Alkondon M, Pereira EF, Castro NG, Schrattenholz A, Barbosa CT, Bonfante-Cabarcas R, Aracava Y, Eisenberg HM, Maelicke A 1997 Properties of neuronal nicotinic acetylcholine receptors: pharmacological characterization and modulation of synaptic function. J Pharmacol Exp Ther 280: 1117–1136
Andoh T, Itoh H, Watanabe I, Sasaki T, Higashi T 2001 Mechanisms of modulation of neuronal nicotinic receptors by substance P and OAG. Am J Physiol Cell Physiol 281: C1871–C1880
Zhou XF, Marley PD, Livett BG 1991 Substance P modulates the time course of nicotinic but not muscarinic catecholamine secretion from perfused adrenal glands of rat. Br J Pharmacol 104: 159–165
Kinney HC, Filiano JJ, Sleeper LA, Mandell F, Valdes-Dapena M, White WF 1995 Decreased muscarinic receptor binding in the arcuate nucleus in sudden infant death syndrome. Science 269: 1446–1450
Ozawa Y, Takashima S 2002 Developmental neurotransmitter pathology in the brainstem of sudden infant death syndrome: a review and sleep position. Forensic Sci Int 14: S53–S59
Author information
Authors and Affiliations
Corresponding author
Additional information
Supported by the Swedish MRC (04X-2887 and 5234), Swedish Match, Stiftelsen Frimurarna Barnhuset, Sällskapet Barnavård, Stiftelsen Samariten, Marianne and Marcus Wallenbergs Foundation and an Unrestricted Bristol-Myers Squibb Neuroscience Grant.
Rights and permissions
About this article
Cite this article
Berner, J., Ringstedt, T., Brodin, E. et al. Prenatal Exposure to Nicotine Affects Substance P and Preprotachykinin-A mRNA Levels in Newborn Rat. Pediatr Res 64, 621–624 (2008). https://doi.org/10.1203/PDR.0b013e318186e5f5
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/PDR.0b013e318186e5f5
This article is cited by
-
Prenatal nicotine exposure decreases the release of dopamine in the medial frontal cortex and induces atomoxetine-responsive neurobehavioral deficits in mice
Psychopharmacology (2017)
-
Evaluation of cognitive behaviors in young offspring of C57BL/6J mice after gestational nicotine exposure during different time-windows
Psychopharmacology (2013)
