
Abstract
Mutations in multidrug resistance 3 gene (MDR3 or ABCB4) underlie progressive familial intrahepatic cholestasis type 3 (PFIC3), a severe pediatric liver disease progressing to cirrhosis. Abcb4−/− mice exhibit slowly developing hepatic lesions that can be accelerated by feeding a cholic acid (CA)–supplemented diet. We investigated the beneficial effects of a soybean lecithin (L)–supplemented diet in this model of liver disease. Abcb4−/− mice and wild-type (WT) controls were divided in four groups by the diet they were fed: control (C) diet, L-supplemented diet, CA–supplemented diet, and L- and CA-supplemented (L+CA) diet. After 2 wk on these regimens, liver enzymes and bilirubin were measured in serum with bile flow, total bile acids, and cholesterol (CHOL) and phospholipid (PL) concentrations in bile. Ductular hyperplasia, portal fibroblastic cell proliferation, myofibroblast activation, and hepatic fibrosis were quantified on liver sections. Abcb4−/− mice fed the C diet exhibited mild liver damage. CA produced very high elevations of serum liver enzymes and bilirubin with significant bile duct proliferation, peribiliary fibroblast activation, and fibrosis. The L-supplemented diet dramatically mitigated the hepatic damage in CA-supplemented diet animals. We conclude that L is protective against liver disease in Abcb4−/− mice and suggest that it could offer potential benefit in PFIC3.
Similar content being viewed by others

Main
Bile formation and the secretion of endogenous substances are the principal functions of the liver. Bile production is a complex process involving canalicular transporters, including members of the adenosine triphosphate–binding cassette protein family, which are key determinants of biliary secretion (1). Impairment of this system can induce cholestasis, a condition associated with the hepatic accumulation of potentially toxic bile constituents and subsequent cell damage. Mutations in canalicular transporter genes can cause some forms of hereditary cholestatic liver disease including PFIC (2,3). These autosomal cholestatic diseases beginning early in life lead to hepatic fibrosis and cirrhosis. There are at least three PFIC types: PFIC1 due to mutations in the ATP8B1 gene (4); PFIC2 due to mutations in the ABCB11 gene, which encodes for a bile salt export pump (5); and PFIC3 due to mutations in the ABCB4 gene encoding for the canalicular phosphatidylcholine (PPC) translocator (6), which that is responsible for PPC transfer from the inner to the outer leaflet of the canalicular membrane. In contrast to ATP8B1 and ABCB11 diseases, ABCB4-deficient patients manifest high serum gamma-glutamyl transpeptidase (GGT) level, ductular proliferation, and inflammatory infiltration of the portal tracts (2).
Mice with homozygous disruption of the Abcb4 gene are unable to secrete PLs into bile and develop lesions characterized by portal tract inflammation, bile duct proliferation, and portal fibrosis (7–9). These animals can be considered a model for human Abcb4 deficiency, a disease ranging from PFIC3 in infants to adult liver cirrhosis (10). However, the lesions progress slowly in Abcb4-deficient mice probably because mice bile contains 50%–70% of muricholate and is hydrophilic, whereas hydrophobic cholate and deoxycholate are prominent in humans. Feeding cholate to Abcb4−/− mice has been shown to increase the hydrophobicity of the bile salt pool and to worsen the liver pathology, including biliary fibrosis (11), confirming that chronic biliary epithelium damage is partly linked to the action of hydrophobic bile acids, which are not inactivated by micellarization with PL.
Despite the availability of some treatments such as ursodeoxycholic acid, hepatic damage often progresses to liver cirrhosis in children with Abcb4 deficiency (12) and liver transplantation is usually required, emphasizing that the search for new therapeutic agents remains a high priority. Studies from this laboratory have shown that feeding animals L-enriched diets stimulates bile secretion under the basal condition and after hepatic bile salt loading (13,14). L also limits the development of hepatic fibrosis induced in animals by carbon tetrachloride or alcohol (15,16).
These findings led us to propose that an L-supplement diet could be beneficial in cholestatic liver disease. The present study examined the effect of an L-supplemented diet on biliary secretion, serum liver enzymes, and liver pathology in Abcb4−/− mice.
MATERIALS AND METHODS
Animals and experimental design.
Abcb4−/− and Abcb4+/+ male mice (FVB/NJ genetic background) were obtained from Jackson Laboratory (Bar Harbor, ME). After weaning, the animals were divided into four diet groups, each containing five to eight animals receiving the following regimens: C diet; L-supplemented diet; C diet with 0.025% of CA (Calbiochem Corp, San Diego, CA); L-supplemented diet with 0.025% of CA (L+CA). Pilot experiments determined that this low level of CA was sufficient to enhance the phenotype of liver disease. Lipid content of the regimens was represented by 16% triacylglycerols (sunflower seed oil) in the C diet and by 20% L granules (Sigma Chemical Co., St. Louis, MO) in the L-supplemented diet. The L granules contained 78% triacylglycerols and 22% PLs. The percentage of triglyceride in both diets was comparable, and the fatty acid composition did not differ significantly between diets. The composition of the C and L-supplemented diets has been described elsewhere (13,14). The mice were housed under a 12:12-h light-dark cycle and with free access to food and drinking water. All protocols were in full compliance with the Université de Montreal guidelines for animal care.
After 2 wk of dietary intervention, the animals were weighed and received i.p. bromodeoxyuridine (BrdU) (4 mg/kg body weight (BW). One hour later, they were anesthetized with i.p. ketamine (4 mg/kg BW) and i.p. xylazine (11.3 mg/kg BW), the abdomen was opened, and the common bile duct cannulated with a PE-10 polyethylene catheter after gallbladder ligation. Body temperature was maintained at 37°C using a thermostatically controlled infrared lamp. Bile was collected (after 3 h of fasting) on ice in preweighed tubes for 1 h in 15-min aliquots and frozen immediately. Bile flow was quantified by weighing the tubes after collection, assuming a density of 1 g/mL. Blood was sampled by aortic puncture, and serum was obtained after centrifugation and stored at −80°C until analysis. After excision, a piece of fresh liver tissue was fixed in 10% neutral formalin and embedded in paraffin for light microscopy; another piece was immediately frozen in liquid nitrogen-cooled isopentane and stored at −80°C.
Serum bilirubin and liver enzymes.
Alanine aminotransferase (ALAT), alkaline phosphatase (ALP), GGT, and bilirubin were studied by routine clinical biochemistry.
Histology, Sirius red staining, immunohistochemistry, and immunofluorescence staining.
Liver tissue sections 5 μm thick were stained with hematoxylin-eosin for routine histology and with Sirius red [saturated picric acid in distilled water containing 0.1% (wt/vol) Sirius red (BDH Chemicals Ltd., Poole, UK)] for visualization of liver fibrosis (17). All slides were mounted with EUKITT (O. Kindler GmbH, Freiburg, Germany). All samples from a series of experiments were stained simultaneously.
To detect α-smooth muscle actin (SMA) expressed by myofibroblasts, paraffin sections were deparaffinized, hydrated, washed with Tris-buffered saline, and incubated with 3% H2O2 in methanol to inhibit endogenous peroxidase. The sections were then incubated with a mouse monoclonal antibody (IgG2a) against SMA (DakoCytomation, Glostrup, Denmark) diluted 1/250 with Tris-buffered saline for 30 min. After washing, the epitopes were detected with the Envision+ system HRP detection kit (DakoCytomation) for 30 min, and the sections were treated with 1 mg/mL diaminobenzidine and 0.1% H2O2 in 0.05 mol/L Tris-HCl (pH 7.6) for about 20 min. After washing with water, the sections were counterstained with hematoxylin and mounted with EUKITT.
For BrdU detection, sections were deparaffinized, hydrated, and treated with 3% H2O2 in methanol and then with 20% human serum containing 1% bovine serum albumin (BSA), with 2 mol/L HCl for 1 h. The sections were incubated overnight at 4°C with a rat antibody against BrdU [diluted 1/500 in phosphate-buffered saline (PBS)/1% BSA; Oxford Technology, Oxford, UK], followed by a rabbit anti-rat antibody (diluted 1/100 in PBS/1% BSA; DakoCytomation) for 45 min. After washing, the epitopes were detected with the Envision+ system HRP detection kit and revealed with liquid diaminobenzidine (DakoCytomation).
To detect cytokeratin expression in bile duct epithelial cells, frozen sections were air-dried, fixed for 10 min in acetone at −20°C, treated with 20% human serum in PBS/1% BSA, and rinsed in PBS. They were then incubated for 45 min with a rabbit polyclonal antibody directed against biliary epithelium specific pancytokeratin (Wide Spectrum Screening; DakoCytomation) diluted 1/800 in PBS containing 1% BSA. After washing, the sections were incubated with a goat anti-rabbit antibody conjugated to Alexa Red (diluted 1/200; Molecular Probes, Eugene, OR) for 45 min. After processing in antifade mounting medium, the sections were examined with a Zeiss Axioplan 2 microscope equipped with epi-illumination and specific filters (Carl Zeiss Microscopy, Jena, Germany). Images were acquired by means of the AxioVision image processing and analysis system (Carl Zeiss Vision). Quantitative data were obtained with a computerized image analysis system (KS 300, Carl Zeiss Vision).
Fibrosis deposition evaluated by the Sirius red staining, bile duct epithelial cell proliferation (cytokeratin staining), and myofibroblastic differentiation (SMA staining) were expressed as a percentage of stained areas on the total measured area. Cellular proliferation was evaluated by the number of BrdU-stained fibroblastic cells present in portal areas. Hepatocytes or biliary epithelial cells stained with BrdU were not included.
All histopathological studies were performed by the same examiner (T.L.), who was blinded to the genetic and dietary status of the mice.
Bile acid determinations in bile.
Total bile acids were assessed with 3α-hydroxysteroid dehydrogenase (Sigma Chemical Co.) (18). Bile acids were identified and quantified by gas chromatography-mass spectrometry in a Hewlett-Packard 5896 gas chromatograph equipped with a Hewlett-Packard 5971A mass selective detector in selected ion monitoring mode. A correction factor was obtained for quantification by using 5β-cholanic acid as internal standard (19).
Lipids in bile.
Cholesterol (CHOL) and PL concentrations were measured after lipid extraction according to Bligh and Dyer (20). Both parameters were quantified enzymatically with commercial kits.
Statistical analysis.
Mean values were compared using three-way ANOVA. When positive, the significance of differences between two groups was evaluated by t test; p < 0.05 was considered significant.
RESULTS
BW and liver weight.
The diets were well accepted by all animals, and the quantity of ingested diet was similar in the different groups. BW gains were similar after 2 wk of experimentation in all WT groups. In Abcb4−/− mice, BW did not change on the C or L-supplemented diet alone. However, the CA-supplemented diet group exhibited a 9.4% BW loss, which was counteracted by L supplementation (L+CA) (data not shown).
The liver weight/100 g BW of all WT groups was unchanged. In knockout (KO) mice, liver weight was significantly increased in the CA-supplemented diet group compared with the C diet group (9.01 ± 0.48 g/100 g BW versus 5.37 ± 2.15 g/100 g BW, respectively; p ≤ 0.05). When these animals also received the L+CA-supplemented diet, liver weight was markedly reduced (5.38 ± 0.45 g/100 g BW) and remained similar to that of the C diet group.
Serum liver enzymes and bilirubin.
Liver enzymes and bilirubin in serum were within the normal range in the four diet groups of WT mice (Fig. 1).

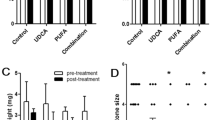
Serum liver enzymes and bilirubin in WT (A) and Abcb4−/− (B) mice. Liver enzymes and bilirubin were significantly increased in ABCB4−/− mice fed the CA-supplemented diet when compared with C diet and L-supplemented diet groups. These changes were dramatically improved by adding L to the diet. C diet (n = 5); L-supplemented diet (n = 6); C diet supplemented with CA (n = 8); L+CA (n = 8). *p < 0.05 compared with the C diet of WT mice, **p < 0.05 compared with C diet of Abcb4−/− mice, †p < 0.05 or ‡p < 0.01 compared with C diet, §p <0.01 when compared with CA-supplemented diet.
In Abcb4−/− mice fed the C diet, serum liver enzymes and bilirubin were significantly higher than in WT mice fed the C diet (Fig. 1). The L-supplemented diet group had decreased bilirubin and GGT levels compared with the C diet group, whereas ALAT and ALP were approximately unchanged. The CA-supplemented diet group showed markedly increased liver enzymes and bilirubin compared with the C diet group. L prevented these elevations with values of L+CA-supplemented diet group being comparable with those of the corresponding controls without CA (L-supplemented diet group).
Bile flow and biliary components.
In WT mice, bile flow and the biliary secretion of total bile acids, CHOL, and PL increased significantly after the addition of CA or L compared with the C diet (Fig. 2). Mice fed the L+CA diet had values that were even more prominent than when these agents were given individually.

Bile flow and secretion of biliary components in WT (A) and Abcb4−/− (B) mice. In WT, CA increased bile flow and secretion of biliary bile acids and lipids, which were even more increased by the addition of L. L did not change bile flow and bile acid secretion significantly in Abcb4−/− mice fed the CA-supplement diet. C diet (n = 5), L-supplemented diet (n = 6), C diet supplemented with CA (n = 8), L+CA diet (n = 8). *p < 0.05, **p < 0.01 or ***p < 0.001 when compared with C diet; †p < 0.01, ‡p < 0.001 when compared with C diet; §p < 0.05, ∥p < 0.01 or ¶p < 0.001 when compared with CA-supplemented diet.
PL and CHOL remained markedly impaired in all Abcb4−/− mice, except in the L-supplemented diet group, which showed significant elevations of CHOL output. In contrast to WT mice, no increase in bile flow and secretion of total bile acids was observed in KO mice after CA feeding. The addition of L (L+CA-supplemented diet) had no significant effect on bile flow and bile acid secretion (Fig. 2).
The results of bile salt composition (data not shown) indicate as expected that in Abcb4−/− mice fed the C diet, the major bile acids were muricholic acids (55 ± 3.6%) followed by CA (43 ± 3.6%). Dihydroxylated bile acids (deoxycholic acid, chenodeoxycholic acid, and ursodeoxycholic acid) contributed 2.25 ± 0.06%. In mice receiving CA, the percentage of contribution of CA increased to 77 ± 7.7% and the contribution of muricholic acids decreased to 18 ± 10%. There was no change in the percentage of contribution of dihydroxylated bile acids. In mice fed both CA and L, CA was the major bile acid (90 ± 2.6%), and the contribution of muricholates was 8.0 ± 2.6%, whereas that of dihydroxylated bile acids was 1.31 ± 0.23% of the total bile acids. Thus, CA was the major biliary bile acid in mice fed the C diet or the L+CA-supplemented diet.
Ductular hyperplasia, myofibroblast activation, cell proliferation in portal areas, and hepatic fibrosis in WT and KO mice.
Standard histology revealed normal portal areas in the four Abcb4+/+ groups, including animals receiving CA (data not shown). Cytokeratin immunostaining disclosed normal biliary structures, no proliferating fibroblastic cells, and no myofibroblast activation in the portal areas. Fibrosis deposition was absent.
The data on Abcb4−/− mice are presented in Figures 3 and 4. In the C diet group, there was only slight damage of the biliary epithelium, which was pseudo-stratified at some places. Cytokeratin immunostaining revealed a normal number of biliary structures. BrdU staining disclosed few proliferating fibroblastic cells in the portal areas (data not shown). SMA immunostaining showed that no myofibroblast activation occurred in the portal areas. Fibrosis deposition, assessed by Sirius red staining, was nearly absent. Histological findings were similar in the L-supplemented diet group.

Standard histology, immunostaining for cytokeratin (CK) and α-smooth muscle actin (SMA), and Sirius red (SR) staining in Abcb4−/− mice. In CA-fed mice (CA), portal tracts were enlarged (HES) with expansion of biliary ductular structures at their periphery (CK), numerous activated myofibroblasts (SMA), and fibrosis deposition (SR). L dramatically improved this hepatic damage in L+CA-diet mice in which histologic findings remained similar to those of the C-diet and L-supplemented diet groups (C and L). Magnification ×100.

Quantification of ductular hyperplasia, portal fibroblastic cell proliferation, myofibroblast activation, and hepatic fibrosis in Abcb4−/− mice. Ductular hyperplasia (CK), myofibroblast activation (SMA), and hepatic fibrosis were assessed by image analyzing and results are expressed as a percentage of stained areas on the total measured area. Portal fibroblastic cell proliferation was determined by counting the number of BrdU-stained cells, and results are expressed as mean ± standard deviation. Quantification confirmed the increase of ductular hyperplasia, myofibroblast activation, fibroblastic cell proliferation, and fibrosis deposition in CA-fed mice, and improvement with L (L+CA). C diet (n = 5), L-supplemented diet (n = 6), C diet supplemented with CA (n = 8), L+CA diet (n = 8).*p < 0.05 when compared with C diet; †p < 0.05 when compared with CA-supplemented diet.
In mice fed a CA-supplemented diet, the portal tracts were enlarged with a significant increment of the number of biliary ductular structures at their periphery. BrdU staining revealed an increase in proliferating fibroblastic cells within the portal areas (data not shown), and numerous SMA-positive myofibroblasts were present, particularly at the periphery of the portal tracts around ductular structures. Fibrosis deposition was significantly increased in the portal areas and within septa.
L dramatically improved the hepatic damage in the L+CA diet group. Fibroblastic cell proliferation and myofibroblast activation even remained similar to that in the corresponding C diet group. No significant fibrosis deposition was noted.
Quantification by image analysis confirmed the increase in ductular structures, fibroblastic cell proliferation, myofibroblast activation, and fibrosis in the CA group. The values remained similar to those of the controls when these mice also received L (L+CA group) (Fig. 4).
DISCUSSION
The Abcb4−/− mouse represents a well-described model of cholangiopathy and biliary fibrosis (7–9) resembling the human liver disease caused by ABCB4 deficiency, which includes a wide spectrum ranging from progressive neonatal cholestasis (PFIC3) to adult liver disease (10). Soon after weaning, Abcb4−/− mice fed normal (C) diet exhibit minimal hepatic pathology that progresses slowly (7). The present studies showed that diets enriched with as low as 0.025% CA are sufficient to dramatically increase serum liver enzymes and bilirubin as well as hepatic fibrosis. CA and other hydrophobic bile acids are known to accumulate in the liver of chronic liver disease patients (21) and worsen the histologic abnormalities (22). Their toxicity has been demonstrated in vivo and in vitro on hepatocytes (23), and they are also known to induce hepatocyte apoptosis (24). Hydrophobic bile acids have also been reported to evoke proliferation of the biliary epithelium in vivo (25) and of cultured cholangiocytes (26). Also, taurocholate has been found to stimulate the secretion of monocyte chemotactic protein-1 and IL-6 (26), two cytokines that play a major role in hepatic fibrogenesis by leading to the recruitment and activation of hepatic stellate cells (27,28). Recently, Fickert et al. (8) demonstrated that bile duct structure alteration is an early step in the pathological process in the Abcb4−/− mice model, leading to bile acid regurgitation from the biliary lumen into the periportal tissue. Subsequent dysregulation of pro- and antifibrogenic genes induces periductal inflammation and fibrosis (9).
We pursued our primary goal, which was to identify agents that could limit or prevent the development of the pathology in the Abcb4−/− mice. This is important because treatment options for many chronic fibrogenic liver diseases are limited. Pharmacological approaches, mostly with ursodeoxycholic acid, can improve clinical and serum parameters in PFIC3 patients but demonstrate highly variable effects on histological changes (12). If no improvement occurs, hepatic transplantation is necessary in the first decade. Therefore, the search for additional intervention strategies to prevent liver disease progression remains a high priority. Based on earlier work by us and others (13–16), we tested whether dietary L could be a potential agent of choice. The present study clearly demonstrates the remarkable effect of dietary L in decreasing the severity of the liver pathology in Abcb4−/− mice, particularly after CA feeding. Interestingly, unpublished preliminary experiments indicated that L was also able to counteract progression of liver disease in long-term experiments in this mouse model, which supports its high clinical potential. Future studies should also evaluate whether L is effective against other models of cholestasis to further characterize the generalizability of its positive effect.
The beneficial effects of L may be due either to its influence on biliary secretion or to effects on hepatic fibrogenesis. L markedly increased bile flow, and the secretion rate of bile acid, CHOL, and PL in Abcb4+/+ mice. This confirms our previous studies in rats in which we also found enhanced bile acid pool size (14). L, however, did not significantly alter the canalicular membrane content of proteins involved in lipid transport to bile (14). We have no data on hepatic canalicular transporters expression in this study, and we are not aware of any published work on this parameter. However, improvement of bile secretion, especially PLs secretion, was not evident in Abcb4−/− mice, which raises doubt about the concept that protection of the biliary epithelium from bile acid toxicity is due to the presence of PPC in bile. The main result of our study is the preventive effects of L on CA-induced fibrosis process in Abcb4−/− mice, with a decreased number of activated myofibroblasts in portal areas and decreased deposition of fibrosis. This is in agreement with the results obtained in toxic models of cirrhosis (15,16,29). Several mechanisms of action may account for the effects of L on biliary fibrosis. First, L may modulate portal myofibroblast activation, which is considered a key event in the development of biliary fibrosis (30) and has been shown to play a major role in the progression of liver lesions in the Abcb4−/− model (9). PPC has been found in vitro to decrease hepatic stellate cell activation (31) and to decrease transforming growth factor β–induced collagen mRNA expression by inhibiting p38 mitogen-activated protein kinase in cultured hepatic stellate cells (32). PPC is also able to stimulate the secretion of collagenase (33), which has been shown to decrease in Abcb4−/− mice (9). Second, L may protect cell membranes from lipid peroxidation, known to promote fibrogenesis (34,35). PPC decreases alcohol-induced oxidative stress in the baboon (36) and prevents lipid peroxidation induced by carbon tetrachloride or ethanol in rats (37). Third, L could protect hepatocytes (38) and cholangiocytes (39) through its antiapoptotic properties.
In conclusion, L can counteract liver disease progression in Abcb4−/− mice, opening new treatment options for children with ABCB4 deficiency.
Abbreviations
- ALAT:
-
alanine aminotransferase
- ALP:
-
alkaline phosphatase
- C:
-
control
- CA:
-
cholic acid
- GGT:
-
gamma-glutamyl transpeptidase
- L:
-
soybean lecithin
- MDR3 or ABCB4:
-
multidrug resistance 3 gene
- PFIC3:
-
progressive familial intrahepatic cholestasis type 3
- PLs:
-
phospholipids
- PPC:
-
phosphatidylcholine
References
Kullak-Ublick GA, Beuers U, Paumgartner G 2000 Hepatobiliary transport. J Hepatol 32: 3–18
Jacquemin E, Hadchouel M 1999 Genetic basis of progressive familial intrahepatic cholestasis. J Hepatol 31: 377–381
Harris MJ, Le Couteur DG, Arias IM 2005 Progressive familial intrahepatic cholestasis: genetic disorders of biliary transporters. J Gastroenterol Hepatol 20: 807–817
Bull LN, van Eijk MJ, Pawlikowska L, DeYoung JA, Juijn JA, Liao M, Klomp LW, Lomri N, Berger R, Scharschmidt BF, Knisely AS, Houwen RH, Freimer NB 1998 A gene encoding a P-type ATPase mutated in two forms of hereditary cholestasis. Nat Genet 18: 219–224
Strautnieks SS, Kagalwalla AF, Tanner MS, Knisely AS, Bull L, Freimer N, Kocoshis SA, Gardiner RM, Thompson RJ 1997 Identification of a locus for a progressive familial cholestasis PFIC2 on chromosome 2q24. Am J Hum Genet 61: 630–633
de Vree JM, Jacquemin E, Sturm E, Cresteil D, Bosma PJ, Aten J, Deleuze JF, Desrochers M, Burdelski M, Bernard O, Oude Elferink RP, Hadchouel M 1998 Mutations in the MDR3 gene cause progressive familial intrahepatic cholestasis. Proc Natl Acad Sci U S A 95: 282–287
Smit JJ, Schinkel AH, Oude Elferink RP, Groen AK, Wagenaar E, van Deemter L, Mol CA, Ottenhoff R, van der Lugt NM, van Roon MA 1993 Homozygous disruption of the murine Mdr2 P-glycoprotein gene leads to a complete absence of phospholipid from bile and to liver disease. Cell 75: 451–462
Fickert P, Fuchsbichler A, Wagner M, Zollner G, Kaser A, Tilg H, Krause R, Lammert F, Langner C, Zatloukal K, Marschall HU, Denk H, Trauner M 2004 Regurgitation of bile acids from leaky bile ducts causes sclerosing cholangitis in Mdr2 (Abcb4) knockout mice. Gastroenterology 127: 261–274
Popov Y, Patsenker E, Fickert P, Trauner M, Schuppan D 2005 Mdr2 (Abcb4)−/− mice spontaneously develop severe biliary fibrosis via massive dysregulation of pro- and antifibrogenic genes. J Hepatol 43: 1045–1054
Jacquemin E, de Vree JM, Cresteil D, Sokal EM, Sturm E, Dumont M, Scheffer GL, Paul M, Burdelski M, Bosma PJ, Bernard O, Hadchouel M, Elferink RP 2001 The wide spectrum of multidrug resistance 3 deficiency: from neonatal cholestasis to cirrhosis of adulthood. Gastroenterology 120: 1448–1458
van Nieuwkerk CM, Elferink RP, Groen AK, Ottenhoff R, Tytgat GN, Dingemans KP, van den Bergh Weerman MA, Offerhaus GJ 1996 Effects of Ursodeoxycholate and cholate feeding on liver disease in FVB mice with a disrupted mdr2 P-glycoprotein gene. Gastroenterology 111: 165–171
Jacquemin E, Hermans D, Myara A, Habes D, Debray D, Hadchouel M, Sokal EM, Bernard O 1997 Ursodeoxycholic acid therapy in pediatric patients with progressive familial intrahepatic cholestasis. Hepatology 25: 519–523
LeBlanc MJ, Gavino V, Perea A, Yousef IM, Levy E, Tuchweber B 1998 The role of dietary choline in the beneficial effects of lecithin on the secretion of biliary lipids in rats. Biochim Biophys Acta 1393: 223–234
LeBlanc MJ, Brunet S, Bouchard G, Lamireau T, Yousef IM, Gavino V, Levy E, Tuchweber B 2003 Effects of dietary soybean lecithin on plasma lipid transport and hepatic cholesterol metabolism in rats. J Nutr Biochem 14: 40–48
Aleynik SI, Leo MA, Ma X, Aleynik MK, Lieber CS 1997 Polyenylphosphatidylcholine prevents carbon tetrachloride-induced lipid peroxidation while it attenuates liver fibrosis. J Hepatol 27: 554–561
Lieber CS, Robins SJ, Li J, DeCarli LM, Mak KM, Fasulo JM, Leo MA 1994 Phosphatidylcholine protects against fibrosis and cirrhosis in the baboon. Gastroenterology 106: 152–159
Waldrop FS, Puchtler H 1982 Light microscopic distinction of collagens in hepatic cirrhosis. Histochemistry 74: 487–491
Turley SD, Dietschy JM 1978 Re-evaluation of the 3 alpha-hydroxysteroid dehydrogenase assay for total bile acids in bile. J Lipid Res 19: 924–928
Dionne S, Tuchweber B, Plaa GL, Yousef IM 1994 Phase I and phase II metabolism of lithocholic acid in hepatic acinar zone 3 necrosis. Evaluation in rats by combined radiochromatography and gas-liquid chromatography-mass spectrometry. Biochem Pharmacol 48: 1187–1197
Bligh EG, Dyer WJ 1959 A rapid method of total lipid extraction and purification. Can J Biochem Physiol 37: 911–917
Greim H, Czygan P, Schaffner F, Popper H 1973 Determination of bile acids in needle biopsies. Biochem Med 8: 280–286
Schmucker DL, Ohta M, Kanai S, Sato Y, Kitani K 1990 Hepatic injury induced by bile salts: correlation between biochemical and morphological events. Hepatology 12: 1216–1221
Delzenne NM, Calderon PB, Taper HS, Roberfroid MB 1992 Comparative hepatotoxicity of cholic acid, deoxycholic acid and lithocholic acid in the rat: in vivo and in vitro studies. Toxicol Lett 61: 291–304
Faubion WA, Guicciardi ME, Miyoshi H, Bronk SF, Roberts PJ, Svingen PA, Kaufman SH, Gores GJ 1999 Toxic bile salts induce rodent hepatocyte apoptosis via direct activation of Fas. J Clin Invest 103: 137–145
Alpini G, Glaser SS, Ueno Y, Rodgers R, Phinizy JL, Francis H, Baiochi L, Holcomb LA, Caligiuri A, Le Sage GD 1999 Bile acid feeding induces cholangiocyte proliferation and secretion: evidence for bile acid-regulated ductal secretion. Gastroenterology 116: 179–186
Lamireau T, Zoltowska M, Levy E, Yousef I, Rosenbaum J, Tuchweber B, Desmouliere A 2003 Effects of bile acids on biliary epithelial cells: proliferation, cytotoxicity, and cytokine secretion. Life Sci 72: 1401–1411
Marra F, DeFranco R, Grappone C, Parola M, Milani S, Leonarduzzi G, Pastacaldi S, Wenzel UO, Pinzani M, Dianzani MU, Laffi G, Gentilini P 1999 Expression of monocyte chemotactic protein-1 precedes monocyte recruitment in a rat model of acute liver injury, and is modulated by vitamin E. J Investig Med 47: 66–75
Choi I, Kang HS, Yang Y, Pyun KH 1994 IL-6 induces hepatic inflammation and collagen synthesis in vivo. Clin Exp Immunol 95: 530–535
Ma X, Zhao J, Lieber CS 1996 Polyenylphosphatidylcholine attenuates non-alcoholic hepatic fibrosis and accelerates its regression. J Hepatol 24: 604–613
Tuchweber B, Desmouliere A, Bochaton-Piallat ML, Rubbia-Brandt L, Gabbiani G 1996 Proliferation and phenotypic modulation of portal fibroblasts in the early stages of cholestatic fibrosis in the rat. Lab Invest 74: 265–278
Poniachik J, Baraona E, Zhao J, Lieber CS 1999 Dilinoleoylphosphatidylcholine decreases hepatic stellate cell activation. J Lab Clin Med 133: 342–348
Cao Q, Mak KM, Lieber CS 2002 DLPC decreases TGF-beta1-induced collagen mRNA by inhibiting p38 MAPK in hepatic stellate cells. Am J Physiol Gastrointest Liver Physiol 283: G1051–G1061
Li J, Kim CI, Leo MA, Mak KM, Rojkind M, Lieber CS 1992 Polyunsaturated lecithin prevents acetaldehyde-mediated hepatic collagen accumulation by stimulating collagenase activity in cultured lipocytes. Hepatology 15: 373–381
Parola M, Leonarduzzi G, Biasi F, Albano E, Biocca ME, Poli G, Dianzani MU 1992 Vitamin E dietary supplementation protects against carbon tetrachloride-induced chronic liver damage and cirrhosis. Hepatology 16: 1014–1021
Lee KS, Buck M, Houglum K, Chojkier M 1995 Activation of hepatic stellate cells by TGF alpha and collagen type I is mediated by oxidative stress through c-myb expression. J Clin Invest 96: 2461–2468
Lieber CS, Leo MA, Aleynik SI, Aleynik MK, DeCarli LM 1997 Polyenylphosphatidylcholine decreases alcohol-induced oxidative stress in the baboon. Alcohol Clin Exp Res 21: 375–379
Chwiecko M, Holownia A, Bielawska A, Farbiszewski R 1993 Inhibition of non-enzymatic lipid peroxidation by ‘Essentiale' a drug enriched in phosphatidylcholine in ethanol-induced liver injury. Drug Alcohol Depend 33: 87–93
Mi LJ, Mak KM, Lieber CS 2000 Attenuation of alcohol-induced apoptosis of hepatocytes in rat livers by polyenylphosphatidylcholine (PPC). Alcohol Clin Exp Res 24: 207–212
Komichi D, Tazuma S, Nishioka T, Hyogo H, Une M, Chayama K 2003 Unique inhibition of bile salt-induced apoptosis by lecithins and cytoprotective bile salts in immortalized mouse cholangiocytes. Dig Dis Sci 48: 2315–2322
Acknowledgements
The authors thank Nathalie Senant (INSERM E0362, Université Victor Segalen Bordeaux 2, Bordeaux, France), and Grace Gavino (Université de Montréal) for their technical assistance. They also thank Schohraya Spahis for her expert secretarial support.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Lamireau, T., Bouchard, G., Yousef, I. et al. Dietary Lecithin Protects Against Cholestatic Liver Disease in Cholic Acid–Fed Abcb4− Deficient Mice. Pediatr Res 61, 185–190 (2007). https://doi.org/10.1203/pdr.0b013e31802d7780
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/pdr.0b013e31802d7780
