
Abstract
Classic galactosemia is caused by impaired galactose-1-phosphate uridyltransferase (GALT EC 2.7.712). If discovered and treated within the first days of life, the acute problems of hepatocellular damage, sepsis, and death are prevented. However, chronic problems such as ataxia, tremor, dyspraxic speech, and ovarian failure may occur. To determine whether screening newborns before discharge from the nursery for GALT deficiency is feasible and whether acute and chronic signs could be prevented by earlier intervention, we developed a simplified “breath test.” We quantitated total body oxidation of 13C-D-galactose to 13CO2 in expired air by normal newborns between 2 h and 2 mo of age and compared their results to older children with GALT deficiency. We found no differences in total body galactose oxidation (TBGO) among normal newborns up to 48 h of age, but a 2-fold rise in TBGO developed during their first 2 wk of life. Older children with galactosemia had significantly less oxidative capacity than normal newborns. We conclude that newborn breath testing for total body galactose oxidation is feasible before discharge from nursery. It has potential utility for both preventing acute neonatal toxicity and determining the mechanisms producing long-term complications such as ovarian failure, dyspraxia, ataxia, and tremors.
Similar content being viewed by others
Main
Classic galactosemia is an autosomal recessive disorder caused by a deficiency of galactose-1-phosphate uridyltransferase (GALT EC 2.7.7.12). The biochemical phenotype is characterized by accumulation of galactose, galactose-1-phosphate, and galactitol in cells, and of excess excretion of galactitol and galactose (1–4). The disease incidence is approximately 1:30,000 live births in the United States (2). If untreated in the first 2 wk of life, poor feeding, jaundice, hepatic failure, Escherichia coli sepsis, and death may occur. We have personal experience with families whose newborns had died (http://www.savebabies.org/diseasedescriptions/galactosemia.php, formally known as, “Tyler for Life Foundation” and personal experience).
Despite population-based newborn screening and dietary intervention within 2–3 wk of life, the clinical outcome of patients with classic galactosemia is variable. Long-term complications include premature ovarian failure, dyspraxic speech, short stature, learning disabilities, and cataracts (5–7). In one ex post facto study, outcome was not related to time of treatment (5).
Most states in the United States screen for galactosemia by the Beutler Method, which measures GALT in dried blood on filter paper through enzyme-linked fluorescence of NADPH production from glucose-1-phosphate. Some states use total galactose quantization (8,9). The median time for results to be received by a referring physician from public health laboratories is poorly documented nationally but from personal experience varies between 4 and 30 d (9).
It is not clear whether this prolonged exposure to excess galactose-1-phosphate produces the chronic effects of galactosemia during fetal development, early infancy, or through prolonged exposure (5). It is recognized that endogenous production of galactose is high in infancy (10,11) and that elevated galactose-1-phosphate remains elevated throughout the galactosemic's life. We have studied and published the toxic effects of elevated galactose-1-P that interfere with UDP glucose production, alter glycoprotein production, and trigger a severe endoplasmic reticulum stress reaction in yeast models and in isogenic human cells cultured from galactosemic patients (12,13).
It is both clinically and scientifically recognized that accumulation of galactose-1-P and possibly other analytes such as galactitol cause acute toxicity in GALT deficiency. By comparison to GALT deficiency, galactokinase deficiency elevates galactitol (but not galactose -1-P) is associated with cataracts but not hepatocellular or ovarian and cerebellar dysfunction (1,2). Thus, there are both fundamental and clinical evidences for the organ specific toxic effects of accumulated galactose-1-P (1,2,5–7,12,13).
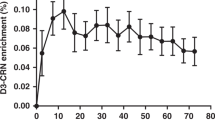
The 13C galactose breath test is safe in normal and galactosemic children and provides a rapid and accurate assessment of total body galactose oxidation (TBGO) (6,7,14,15). This breath test also has a high predictive value for dyspraxia and ovarian failure in older galactosemic patients (6,7). We previously demonstrated that the cumulative percent dose recovered as 13C enrichment of CO2 gives equivalent results whether 13C-galactose was administered orally or intravenously (14,15), thus enabling the potential for breath test screening in normal newborns before discharge from the nursery.
In this study, we developed and piloted validation of a noninvasive oral galactose breath test. We used this method to analyze TBGO to expired CO2 in newborns from 2 to 48 h of age and repeated these studies in the same normal infants at 2, 4, and 8 wk of age. We compared these results with older galactosemic babies and children of known GALT genotype who are being treated with galactose-restricted diets.
METHODS
Patients.
Simplified “breath tests” were completed on 21 normal newborns (including one pair of twins) and 18 babies and children diagnosed with galactosemia. Normal babies and the galactosemic children were studied at the General Clinical Research Center (GCRC), at Jackson Memorial Hospital and at the Mailman Center for Child Development, University of Miami, Miller School of Medicine in Miami, Florida. Some children with galactosemia were studied at the Parents for Galactosemic Children (PGC) meeting in Philadelphia, as a field trial. All studies were approved by the Western Institutional Review Board (WIRB, Olympia, WA) and with the informed consents of parents. The 21 normal newborns were the children of healthy pregnant women who understood and signed consents in either Spanish or English before delivery in accordance with the IRB regulations. Eleven of them were studied immediately following elective cesarean sections. The criteria for entry of normal newborns included normal weight, gestational age greater than 37 wk, and a lack of pregnancy or delivery complications. The 18 babies and children, age 2 wk to 140 mo, with classic galactosemia, were tested once and compared with normal newborns.
Breath test.
The breath from all babies and infants were quantitated for 13CO2 enrichment in expired air before and after an oral administration of 7 mg/kg of an aqueous solution of D-[1-13C]-Galactose (from a GMP-supervised D-galactose powder, Omicron Biochemicals, Inc., South Bend, IN) delivered by a syringe in a volume of 2.5–5 mL of aseptically filled 10 mg/mL solution into the infants buccal fold. After baseline breath collection, test samples were collected at 30, 45, 90, 100, 110, and 120 min time points (post dose) using a face mask with one-way inlet and outlet valves connected to the standard collecting bag in newborns. Their ages at encounters were at 2–4 h, 12–24 h, and 36–48 h after birth and then again at 2 wk and 1 and 2 mo of age. Information regarding activity, feeding, complications, and demographics were recorded at each time point. In galactosemic infants and children, breath was collected by either face mask or by blowing into breath collection test tubes with a straw. Duplicate samples (field trial at PGC conference) were collected only at baseline and at 2 h and then transferred to four 10-mL vacuum tubes that were devoid of 13C and stored at room temperature until analysis of 13CO2 enrichment.
Measurement of 13CO2 in the air.
In each duplicate breath sample, the molar ratios of 13CO2 to 12CO2 were quantitated by a gas isotope ratio mass spectrometer (GIRMS) (EUROPA Automated breath carbon analyzer). A delta over the baseline (DOB) was calculated from the 13CO2/12CO2 from the following equation: DOB = [RT – RB /RS] × 1000, where RT is the post dose sample, RB is baseline and RS is the reference standard, Pee Dee Belemnite. To determine production rates of CO2 (mmol CO2/min) in expired air, basic metabolic rate (BMR), and energy expenditure (EE) were calculated from estimates based on the Schofield equation from age, sex, height, and weight. Energy expenditure, O2 consumption, and CO2 production were calculated from the Schofield and de Weir equations (16,17). Note that these constants were derived from older children.
The cumulative percent dose of ingested 13C-D-galactose retrieved as 13CO2 in expired air (CUMPD) was calculated for the area under the curve by trapezoidal integration.
CUMPD = i = n Σi = 1 (PCDi + PCDi –1) (ti – ti –1)/2, where PCDi and ti represent the percentage dose in mmol/min and time in minutes at the ith breath collection point.
Thus, the DOB was a direct measurement of 13CO2 enrichment and the CUMPD was derived from the DOB assuming constants from metabolic rates, energy expenditure, and rates of CO2 production (16,17). We collected duplicate samples of breath before and over 120 min after D-[1-13C]-galactose ingestion.
Statistical analysis.
Continuous data were reported as means ± SD. Paired t test and independent samples t test and one-way ANOVA were used to test the differences between the CUMPD obtained at 120 min values for the same patient, between normal newborns and older children with classic galactosemia, and among newborns of different age groups, respectively. We used a receiver operating characteristic (ROC) analysis to determine a cut-off value of CUMPD that would provide a “positive” breath test for galactosemia. For a cut-off value of CUMPD at 120 min of 1.17%, there was a sensitivity of 0.97 and a specificity of 0.96. However, because of relatively small numbers, the area under the curve of 0.97 failed to reach significance.
RESULTS
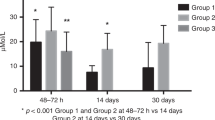
Breath tests were conducted in 21 healthy babies, and 6 completed longitudinal studies from birth to 2 mo (Table 1). To determine whether significant changes in total body oxidation of galactose occurred within 2 d of life, we compared results within the first 48 h of life (Table 1). Eighteen normal newborns were studied at 2–4, 12–24, and 36–48 h of age. Eight studied at 2–4 h of life had a mean CUMPD at 120 min of 4.33 ± 3.66% (range, 0.64–10.81). Thirteen normal newborns studied at 12–24 h of life had a CUMPD value at 120 min of 5.78 ± 2.44% (ranges, 1.07–9.51). Fourteen normal newborns studied at age 36–48 h had a CUMPD value at 120 min of 4.40 ± 2.11% (ranges, 1.01–8.45) (Table 1). Thus, there were no differences in galactose oxidation to CO2 within the first 2 d of life from age 2 h to age 48 h (p < 0.01).
By contrast, there was an increase in TBGO from birth to 2 wk of age among the normal newborns (Table 1). To examine this increase in the oxidative capacity, six normal newborns volunteered for long-term follow up after discharge from the nursery (Tables 1 and 2). The breath tests results were compared in the same infants at <24 h of age and at 2 mo of age. At birth their mean CUMPD was 5.39 ± 1.65% and it increased at 2 mo of age to 10.69 ± 3.61%, p = 0.05 (Table 2). Thus, there was a 2-fold increase in total body galactose oxidation to CO2 that developed in normal neonates between 2 d and 1 mo of life.
To compare the results of breath testing in normal newborns to children with galactosemia, we studied 18 babies and children with known galactosemia who were being treated with lactose-restricted diets. Their genotypes and ages are presented in Table 3. The mean values of CUMPD were lower in these older treated galactosemic children than in normal newborns at all ages (compare Tables 1 and 3). The intraindividual CUMPD among newborns had some expected intraindividual variation due to unknown circumstances such as swallowing, regurgitation, gastric emptying, etc. (Table 1). There were three overlapping results among 44 breath tests in normal newborns 2 wk of age or less (NNB 01, 04, and 05 in Table 1) with 2 of the 18 tests of the treated older children with galactosemia (GA01 and 10 in Table 3). However, the mean among all 55 studies of normal TBGO was statistically different from all galactosemic children regardless of age. The value of CUMPD in galactosemic children was lower than that for 14 normal children at age 2 d (4.40 ± 2.11) p < 0.001 and normal children at 2 wk of age (9.95 ± 5.09) p < 0.001 (Table 4). The 18 children with galactosemia had a mean CUMPD of 0.39 ± 0.41 (Table 4). Therefore, older galactosemic children who now have lower total body galactose pools than during their newborn period still have statistically less galactose oxidation than normal newborns. We attempted validation studies using the receiver operating characteristic statistic. Using the 120-min CUMPD of 1.17% as a cut-off value, sensitivity and specificity were 0.97 and 0.96, respectively. The sample size was not large enough for statistical significance to determine whether this level of galactose oxidation in newborns could be used as a discriminant for galactosemia screening before discharge from the nursery, but was a highly suggestive cut-off number.
DISCUSSION
We previously studied TBGO by older patients with galactosemia and found that a single oral bolus of 7 mg/kg of D-[1-13C]-galactose was safe and correlated with previous, more invasive intravenous studies (14). Neither RBC gal-1-P nor urine galactitol increased within 24 h of testing. Other metabolic disorders that express a serious problem in newborns have been studied following bolus or sustained exposure to trace amount of precursors in blocked reactions (18,19). Children with maple syrup urine disease and phenylketonuria had no adverse events to bolus or sustained infusions of leucine or phenylalanine respectively at these trace doses for 13C detection in breath. Therefore, application of this noninvasive oral “breath test” to infants with or without galactosemia seemed a reasonable and ethical endeavor.
It should be noted that enrichment of CO2 with 13C precursor carbon depends on the total body pool of the unlabeled precursor. Therefore, the treated older child with galactosemia would have a lower pool of unlabeled galactose than when untreated during early infancy of life. Therefore, their TBGO should be lower during early infancy than later after lactose restriction and maturation of their galactose oxidative pathways.
In fact, our previous studies of normal and galactosemic adults and older children for TBGO gave a cut off of 5% CUMPD that was significant to discriminate outcomes of GALT deficiency (6,7,14). Endogenous production of galactose by normal and galactosemic patients is quantified in g/kg/d and thus newborns with GALT deficiency are particularly stressed (10). The older children with galactosemia whose galactose intakes were restricted, whose TBGOs were mature, and whose galactose pools were minimal, still had lower TBGO than normal infants.
There are little data to support the scientific notion that energy expenditure and basic metabolic rate are the same in infants and in older children (16,17). Previous studies using an infant respiratory chamber directly quantitated energy expenditure and basal metabolic rates in infants from 3 to 4 mo of age and found some differences from World Health Organization figures, particularly in those with HIV or other causes of growth retardation (20,21). When measured over longer times (24 h) the energy expenditure in active infants was underestimated by the Schofield equation (16,20,21). However, there were no differences between inferred and directly measured resting metabolic rates (21). We made the assumptions that the data from Schofield and Wier equations could be used to determine the derived CUMPD from the raw data of 13CO2 enrichment over baseline (DOB).
Our data supports the surprising finding of an increase in TBGO to CO2 by normal infants between the ages of 2 d and 2 wk. The percentage of labeled galactose recovered in expired air doubled from 4.40% to 9.96% at 2 wk and remained increased at 1 and 2 mo of age (Tables 1 and 2). There is precedent for the development of increased activity during normal infant development for important metabolic pathways. Perhaps, the best studied is UDP-glucuronyl transferase I (UGTA-1) and the ability of normal newborns to develop mechanisms for conjugating bilirubin (22,23). There is a relationship between plasma bilirubin concentration and TA repeats in the promoter region of the UGTA-1 gene such that heterozygotes and homozygotes with longer TA repeats have a significantly higher concentration of bilirubin in the newborn period than those with fewer repeats (23). The continuum from normal neonatal hyperbilirubinemia response to phenobarbital and Gilbert's syndrome is a paradigm to explain our observations. Total body oxidation of galactose to CO2 increased over the first weeks of life. The GALT enzyme is rate limiting in this process and its gene has a promoter region with at least one defined positive response element that includes three sequential GTCA repeats in a carbohydrate response domain (24,25). Little is known in humans concerning the developmental control of helix/loop/helix trans acting proteins that might control the expression of GALT through this carbohydrate response domain. GALT is the rate-limiting enzyme in TBGO and its developmental control would be of general scientific interest in this evolutionarily conserved metabolic pathway (25).
Regardless of the fundamental mechanisms involved in increasing the normal infant's ability to oxidize galactose, our data indicated that a simplified and a noninvasive breath test could differentiate children with GALT deficiency from normal newborns. TBGO by older children with galactosemia was an order of magnitude lower than normal infants (Tables 1, 3, and 4). The mean CUMPD of 4.40% of 13CO2 recovered in normal newborns at 2 d of age is clearly above the CUMPD of 0.42% recovered from older treated children with GALT deficiency.
We had previously achieved several objectives in developing a suitable, noninvasive breath test for neonates. The 13C-D-galactose was delivered orally with safely. Mutations in GALT clearly reduced 13CO2 recovery in expired air. The breath test predicted the severity of disease and the genotype of GALT (6,7,14). We have added another dimension in the current studies. We have successfully adapted the galactose breath test to the normal neonate and have identified the need for age-specific normal data. The routine use of this simple “breath test” as a screen for galactosemia before discharge from the nursery might save the lives of newborns who have classic galactosemia and are breast-feeding while awaiting results of traditional population based, newborn screening methods. One could visualize a nurse collecting a baseline breath giving 2 mL of C13 galactose by mouth to up to 30 newborns, collecting a breath sample 90–120 min later and analyzing it on an infrared or cycloidal mass spectrometer in the nursery before discharge. If less than 2% of 13C were recovered in the breath, the babies would be given soy-based formula until the state newborn screen results were returned. With this scenario, no blood is necessary and a nurse or nurse-based technician could perform the entire screening, much like hearing tests are now administered before discharge.
Early diagnosis and intervention before neurologic or hepatocellular damage occurs could also answer the cause(s) for poor outcomes in patients with classic galactosemia. Are ovarian failure, dyspraxic speech, tremor, growth restriction, or ataxia caused in utero or during the neonatal exposure to combined endogenous production and exogenous ingestion of galactose? Perhaps reducing this toxic load during the first days of life could answer this question.
Several barriers still exist. The gas isotope ratio mass spectrometer is expensive and will not be a suitable instrument in a nursery laboratory. However, several much less expensive instruments are available such as C13/C12 specific cycloidal mass spectrometer and CO2 gas sensors based on infrared chemistry. Vernier CO2 gas sensors are used to monitor gaseous carbon dioxide levels in chemistry experiments and the Japanese Company Otsuka Electronics, Inc. developed the UBiT infrared analyzer. The UBiT can quantitate 13CO2 in breath and is used to detect Helicobacter pylori when 13C-labeled urea is converted by this organism's urease to 13CO2 in breath. A difficulty with these less expensive technologies is low sensitivity (requires one in 300–5000 ppm of CO2). Our samples contained one in 10,000–20,000 ppm) and thus up to 250 mL of expired air would need to be analyzed. These are not insurmountable problems and comparative field trials are underway with this type of instrument (27).
Abbreviations
- CUMPD:
-
cumulative percent dose of ingested 13C-D-galactose retrieved as 13CO2 in expired air
- DOB:
-
delta over the baseline
- GALT:
-
galactose-1-phosphate uridyltransferase (EC 2.7.7.12)
- TBGO:
-
total body galactose oxidation
References
Holton JB, Walter JH, Tyfield LA 2001 The Metabolic and Molecular Bases of Inherited Diseases, 8th Ed. McGraw Hill, New York, pp 1533–1587
Elsas LJ 2000 Galactosemia. GeneReviews, University of Washington. Retrieved from: http://www.geneclinics.org. Accessed on March 15. 2005
Leslie ND, Immerman EB, Flach JE, Florez M, Fridovich-Keil JK, Elsas LJ 1992 The human galactose-1-phosphate uridyl transferase gene. Genomics 14: 474–480
Elsas LJ, Lai K 1998 The molecular biology of galactosemia. Genet Med 1: 40–48
Waggoner DD, Buist NR, Donnell GN 1990 Long-term prognosis in galactosemia: results of a survey of 350 cases. J Inherit Metab Dis 13: 802–818
Guerrero NV, Singh RH, Manatunga A, Berry GT, Steiner RD, Elsas LJ 2000 Risk factors for premature ovarian failure in females with galactosemia. J Pediatr 137: 833–841
Webb AL, Singh RH, Kennedy MJ, Elsas LJ 2003 Verbal dyspraxia and galactosemia. Pediatr Res 53: 396–402
Beutler E, Baluda M 1966 Biochemical properties of human red blood cell galactose-1-phosphate uridyl transferase (UDP glucose:α-D-galactose-1-phosphate uridyl transferase) from normal and mutant subjects. J Lab Clin Med 67: 947–954
Therrell B 2005 National Newborn Screening and Genetics Resource Center. Retrieved from: http://genes-r-us.uthscsa.edu. Accessed on June 27, 2007
Berry GT, Nissim I, Lin Z, Mazur AT, Gibson JB, Segal S 1995 Endogenous synthesis of galactose in normal men and patients with hereditary galactosemia. Lancet 346: 1073–1074
Schadewaldt P, Kamalanathan L, Hammen HW, Wendel U 2004 Age dependence of endogenous galactose formation in Q188R homozygous galactosemic patients. Mol Genet Metab 81: 31–44
Lai K, Langley SD, Khwaja FW, Schmitt EW, Elsas LJ 2003 GALT deficiency causes UDP-hexose deficit in human galactosemic cells. Glycobiology 13: 285–294
Slepak T, Tang M, Addo F, Lai K 2005 Intracellular galactose-1-phosphate accumulation leads to environmental stress response in yeast model. Mol Genet Metab 86: 360–371
Berry GT, Singh RH, Mazur AT, Guerrero N, Kennedy MJ, Chen J, Reynolds R, Palmieri MJ, Klein PD, Segal S, Elsas LJ 2000 Galactose breath testing distinguishes variant and severe galactose-1-phosphate uridyltransferase genotypes. Pediatr Res 48: 323–328
Berry GT, Nissim I, Gibson JB, Mazur AT, Lin Z, Elsas LJ, Singh RH, Klein PD, Segal S 1997 Quantitative assessment of whole body galactose metabolism in galactosemic patients. Eur J Pediatr 156: S43–S49
Schofield WN 1985 Predicting basal metabolic rate, new standards and review of previous work. Hum Nutr Clin Nutr 39: 5–41
Weir JB 1990 New methods for calculating metabolic rate with special reference to protein metabolism. Nutrition 6: 213–221
Elsas LJ, Ellerine NP, Klein PD 1993 Practical methods to estimate whole body leucine oxidation in maple syrup urine disease. Pediatr Res 33: 445–451
Treacy EP, Delente JJ, Elkas G, Carter K, Lambert M, Waters PJ, Scriver CR 1997 Analysis of phenylalanine hydroxylase genotypes and hyperphenylalaninemia phenotypes using L-[1-13C]phenylalanine oxidation rates in vivo: a pilot study. Pediatr Res 42: 430–435
Duro D, Rising R, Cole C, Valois S, Cedillo M, Lifshitz F 2002 New equations for calculating the components of energy expenditure in infants. J Pediatr 140: 534–539
Cole CR, Rising R, Hakim A, Danon M, Mehta R, Choudhury S, Sundaresh M, Lifshitz F 1999 Comprehensive assessment of the components of energy expenditure in infants using a new respiratory chamber. J Am Coll Nutr 18: 233–241
Black M, Sherlock S 1970 Treatment of Gilbert's syndrome with phenobarbitone. Lancet 1: 1359–1361
Bosma PJ, Chowdhury JR, Bakker C, Gantla S, de Boer A, Oostra BA, Lindhout D, Tytgat GN, Jansen PL, Oude Elferink RP, Chowdhury NR 1995 The genetic basis of the reduced expression of bilirubin UDP-glucuronyltranserease 1 in Gilbert's syndrome. N Engl J Med 333: 1171–1175
Elsas LJ, Lai K, Saunders CJ, Langley SD 2001 Functional analysis of the human galactose-1-phosphate uridyltransferase promoter in Duarte and LA variant galactosemia. Mol Genet Metab 72: 297–305
Elsas LJ, Webb AL, Langley SD 2002 Characterization of a carbohydrate response element regulating the gene for human galactose-1-phosphate uridyltransferase. Mol Genet Metab 76: 287–296
Lai K, Elsas LJ 2001 Structure–function analyses of a common mutation in blacks with transferase-deficiency galactosemia. Mol Genet Metab 74: 264–272
CO2 Gas Sensor. Retrieved from: http://www.vernier.com/probes/probes.html?co2-bta. Accessed on March 18, 2007
Acknowledgements
The authors thank Helen Travers, M.S., CGC of Genzyme Corporation, Cambridge, MA, for helping coordinate patients, and Advance Breath Diagnostics, LLC for performing quantitative studies of 13CO2 in air. We also thank the Parent's of Galactosemic Children (PGC) for their encouragement and hospitality.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supported by a USPHS from National Institutes of Health R44-DK60054 and MO1RR16587, “Improved Outcome Prediction in Galactosemic Newborns”, and by The Dr. John T. Macdonald Foundation.
Rights and permissions
About this article
Cite this article
Barbouth, D., Velazquez, D., Konopka, S. et al. Screening Newborns for Galactosemia Using Total Body Galactose Oxidation to CO2 in Expired Air. Pediatr Res 62, 720–724 (2007). https://doi.org/10.1203/PDR.0b013e3181598cdf
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/PDR.0b013e3181598cdf
This article is cited by
-
Introduction to the Maastricht workshop: lessons from the past and new directions in galactosemia
Journal of Inherited Metabolic Disease (2011)
