
Abstract
MicroRNAs (miRNAs) regulate protein-coding genes post transcriptionally in higher eukaryotes. Argonaute proteins are important in miRNA regulation and are also implicated in epigenetic mechanisms such as histone modifications and DNA methylation. Here, we review miRNA regulation and outline its connections to epigenetics.
Similar content being viewed by others


Main
The discovery of RNA interference (RNAi)—a molecular process where double-stranded RNA can deplete mRNA via sequence-specific mechanisms—demonstrated both effective and specific RNA-mediated gene silencing (1). When, 3 years later, researchers identified a large class of nonprotein coding RNAs called microRNAs (miRNA), they confirmed the potential for large scale endogenous silencing (2–4). As subsequent research has unraveled these processes, we have seen that miRNAs have become increasingly important to our understanding of gene regulation. Also, miRNAs appear to be involved in many aspects of development, including the establishment and maintenance of tissue-specific expression profiles. Previously defined epigenetic mechanisms, such as DNA methylation and histone modification, are also important modifiers of gene expression. In some species, regulatory RNAs possess epigenetic-like properties, and in vitro experiments have linked RNAi to the classic epigenetic mechanisms, which further emphasize the need to view miRNAs as part of a larger apparatus of regulatory mechanisms.
This review will briefly describe epigenetic mechanisms before introducing important aspects of miRNA regulation. This includes transcription, biogenesis, and targeting, in addition to the relationship of miRNAs to other RNAs that associate with parts of the miRNA pathway. Also, we discuss the various epigenetic traits that RNA regulation possess, including possible links to the classical epigenetic mechanisms, and finally outline the clinical importance of miRNAs going forward.
EPIGENETICS AND THE EPIGENOME
Epigenetics is the study of heritable changes in gene function that cannot be attributed to changes in the DNA coding sequences. For example, the regulatory state of a cell is inherited by its daughter cells following cell division, but the cells' DNA may be identical to that of other cells that do not share the regulatory state. Epigenetic inheritance pertains to both mitotic and meiotic cell divisions. The classical epigenetic mechanisms include DNA methylation and histone modifications, but other mechanisms of gene regulation—especially those that involve nonprotein coding RNA—have become increasingly important. The epigenetic state of the cell, meaning the status of the various epigenetic mechanisms, is often referred to as the epigenome. Note, however, that the biologic end point, which is the regulatory state of the cell, is easier to observe directly than the actual epigenetic modifications.
In the following, we will briefly introduce the classical epigenetic mechanisms. Detailed reviews have been published elsewhere (5–7).
DNA methylation.
As the name suggests, DNA methylation involves the addition of a methyl group to DNA residues. For example, the most extensively studied methylation is that of the fifth carbon of the cytosine's pyrimidine ring—a potentially mutagenic event that frequently causes C:G to T:A transitions. CpG islands—phosphodiester-linked cytosine and guanine pairs that span a region of at least 200 base pairs with more than 55% GC content—are found in approximately 40% of the promoters of mammalian genes. These islands are usually unmethylated when the genes are expressed and vice versa, which spurred the interest in DNA methylation as a general mechanism for transcriptional gene silencing.
In addition to maintaining regulatory roles that are important for the cell's function, DNA methylation plays a role in genome maintenance both in the defense against viral sequences and silencing of transposable elements. When the cell divides, the pattern of DNA methylation is maintained in the daughter cells. DNA methyltransferases—of which there are three types in mammals—are responsible for the DNA methylation. DNA methyltransferases 3a and 3b (Dnmt3a and Dnmt3b) initiate new methylation (8), whereas Dnmt1 is a maintenance enzyme (9). The function of Dnmt2 was undefined for a long time, but it was recently shown that Dnmt2 does not methylate DNA, but instead targets a specific position of aspartic acid tRNAs (10). Thus, Dnmt2 is in fact an RNA methyltransferase.
Histone modification.
Histones are the main proteins of chromatin, which allows DNA to be very condensed in the cell's nucleus. Four histone classes—a H3-H4 tetramer and two H2A-H2B dimers—form the octamer that constitute the core histones around which a little less than two helical turns of DNA's double helix wrap (11). This situation is often schematically illustrated like beads on a string, and the structure folds into higher order chromatin, which is very compact. A gene can only be transcribed if the chromatin structure changes temporarily to allow regulatory proteins to bind the relevant portion of the DNA. Histone tails—short arms that are separate from the main structure—can be acetylated, methylated, phosphorylated, and ubiquitinated to change the histone structure and therefore enable or prevent access to the DNA. For example, acetylation most often marks active regions of transcription, whereas methylation can be present in both active and inactive regions. The vast number of combinations that exist of such histone tail modifications means that there may be a histone code that can be read by the transcriptional apparatus (6).
Potential for RNA-mediated effects.
A number of nonprotein coding RNAs play important roles in modifying the sequence, structure, or expression of mRNAs and thereby also changes the protein expression from these genes. For example, small nuclear RNAs (snRNA) are involved in a range of processes, including gene splicing, telomere maintenance, and transcription factor regulation, whereas small nucleolar RNAs (snoRNA) guide chemical modifications to other RNA genes and guide RNAs (gRNA) are involved in RNA editing. One of the most interesting families of such nonprotein coding RNAs is the main topic of this review, namely the miRNAs that appear to be involved in gene regulation on multiple levels.
Whether regulatory changes are induced by transcription factors or mediated by RNAs, the cells can maintain the expression level long after the original factors that stimulated the effect has vanished. Cells differentiate into various types, but when their expression program has been established, their daughter cells remain of the same type even though the factors that triggered the changes may be gone. We currently have only limited knowledge about how nonprotein coding RNAs in general and miRNAs in particular play into this picture. Also, while it is unclear whether these RNA effects are inherited like epigenetic mechanisms, a recent paper demonstrated the potential for RNA-mediated inheritance (12).
MicroRNA REGULATION
miRNAs are nonprotein coding RNA genes that reside within longer transcripts as distinct hairpins that mature into 22 nucleotide (nt) long sequence-specific gene regulators. Since the initial discoveries that the small RNAs let-7 and lin-4 are crucial for C. elegans' correct transition from larvae into adult worm (13–16), researchers have identified miRNAs as an abundant and highly conserved gene class (2–4). Currently, the online repository of miRNA sequence data, miRBase, contains nearly 500 human miRNAs (474; miRBase release 9.0) (17), but recent reports estimate 1000 or more miRNAs in the human genome (18,19). Although some miRNAs, such as let-7, are conserved in most animals, many of the recently identified human miRNAs are specific to primates (18,20) and some are even human-specific (19). Curiously, the miRNAs identified in the initial small RNA cloning efforts are both highly expressed and widely conserved, whereas the more lineage-specific miRNAs appear to be less abundant—possibly because these newer miRNAs are very cell-specific or expressed at low levels in many different cells and tissues. Conservation of sequence does not, however, necessarily imply conservation of function, as expression patterns for some conserved miRNAs have diverged with evolutionary distance (21).
Transcription.
Although little is known of miRNA regulation and transcription, the best characterized miRNAs originate from independent RNA polymerase II transcripts (22,23) or from introns or exons of protein-coding or nonprotein-coding genes (24). Intragenic miRNAs are relatively common, as 130 of the 464 human miRNAs from miRBase 8.1 map to protein-coding genes in UCSC's genome browser database (25). The majority of these intragenic miRNAs (107 of the 130) are sense transcripts that can be transcribed as part of the host gene, then spliced out, and further processed. Such intronic miRNAs rely on the established transcription and splicing of their host genes and are therefore only present when their host gene is transcribed. In effect, intronic miRNAs represent a simple mechanism for a protein-coding gene to down-regulate other protein-coding genes. Furthermore, mutations that lead to new miRNAs in introns could be an effective mechanism to establish a well-regulated miRNA and thereby be a driving force in animal evolution (26). However miRNAs are regulated and transcribed, they tend to cluster throughout the genome (3,27), and many of these clusters are likely transcribed as polycistrons (28).
Biogenesis.
Post transcription—and splicing for intronic miRNAs—miRNAs rely on at least three different protein complexes to mature into the short ∼22 nt single-stranded gene regulators. First, the nuclear Microprocessor complex, which consists of DGCR8 and the ribonuclease (RNase) III Drosha, processes the primary transcript into miRNA precursors (pre-miRNAs) (29–32). Most known animal miRNAs have a hairpin with a ∼33 base pair stem flanked by a single-stranded region, and current models suggest that DGCR8 recognize this characteristic structure and guides Drosha to cleave the hairpin about 11 nts from the single-stranded region (33). Drosha or other unknown co-factors may, however, also recognize some of the miRNA hairpin's key features and thereby contribute to cleavage specificity (34,35). Second, the carrier protein Exportin-5 and Ran GTPase transport pre-miRNA from the nucleus to the cytoplasm (36–38). Exportin-5 recognizes the precursor stem and the characteristic RNase III 3′ overhang (39) and may represent a rate-limiting step in miRNA biogenesis (40,41). Third, the cytoplasmic RNase III Dicer cleaves the pre-miRNA about 22 nts from its end (42–44). Dicer and the Tar RNA binding protein (TRBP) hands the duplex over to Argonaute 2 (Ago2), which then incorporates one of the duplex strands and cleaves or dissociates the other strand (45–48). In Drosophila, Ago2, with the aid of R2D2 (49), preferentially incorporates the strand that has the least stable 5′ end (50,51), which explains why one of the duplex strands predominates as the mature miRNA. Disruptions in any of these three processing steps can alter miRNA expression patterns (52,53). The mechanism by which the antisense strand is appropriately handed off to Ago 2 in mammals is not clearly defined as of yet.
Targeting.
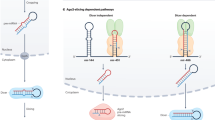
The mature miRNA, bound to Ago2 (54,55), forms the core of the RNA-induced silencing complex (RISC) (56). The miRNA guides RISC to mRNAs that have miRNA-complementary sites and RISC then cleaves (57,58), degrades (59–61), or suppresses translation (14,15) of the target mRNA, depending on the degree of complementarity between miRNA and mRNA (Fig. 1). Cleavage is the strongest mechanism and is also the most specific, as the miRNA and mRNA must form a near-perfect duplex to induce cleavage (62–65). Degradation and translational suppression require less complementarity; seven consecutive base pairs between the mRNA and nucleotides 2–8 from the miRNA's 5′ end are enough to reduce the protein levels of the target (66). Consecutive matches between mRNA and miRNA nucleotides 2–8 or 2–7 are commonly referred to as seed sites, and analyses of conserved seed sites in 3′ untranslated regions (UTR) have shown that a single miRNA may regulate hundreds of genes (66–70). An experiment that over-expressed miR-1 and miR-124 confirmed this potential, as microarray analyses showed that each miRNA down-regulated more than 100 mRNAs (71). Seed sites are not necessary for miRNA targeting, however, as target sites can have mismatches or GU-wobble base pairs within the seed region and still be functional (72,73). To be functional, however, these sites do require more extensive base-pairing between the mRNA and the miRNA 3′ end than do the seed sites (66). Furthermore, even if conserved seed sites are the best current method for predicting miRNA targets, a conserved seed site is not sufficient for down-regulation (74). Instead, miRNA targeting likely relies on the target site's sequence context (72).

Two types of miRNA target sites. (a) Near perfect complementarity between miRNA and mRNA gives mRNA cleavage, whereas (b) less complementarity gives mRNA degradation and translational suppression.
OTHER RNAs THAT ASSOCIATE WITH THE miRNA PATHWAY
As previously mentioned, animal miRNAs are only one of several types of small regulatory RNAs (75). Some of these are similar to miRNAs and associate with several of the protein complexes that are used by animal miRNAs. Plant miRNAs and exogenous and endogenous small interfering RNAs (siRNAs) are the best characterized small RNAs that associate with Dicer, Ago1, and their homologues, but cloning studies have identified several other small RNA that may rely on one or more of these proteins for biogenesis or function. The different classes of small RNAs have been implicated in many different mechanisms (Table 1).
Plants, like animals, have 22 nt RNAs that mature from hairpins within longer primary transcripts in a series of distinct processing steps (76–78). Because of the similarities to animal miRNAs, these RNAs were also named miRNAs, but the processing pathways and dominant targeting mechanisms for animal and plant miRNAs are so different that animal and plant miRNAs can be considered two different gene classes. The differences between animal and plant miRNAs can be summarized as follows. First, plants lack a Drosha homologue, and so a Dicer homologue processes the primary transcript into a duplex form in the nucleus (79). This lack of Drosha processing likely explains why plant miRNA hairpin stems do not have the animal miRNAs' well-defined length of about three helical turns, but vary from 60 to more than 400 nucleotides (miRBase 8.2). Second, the ends of plant miRNAs are methylated, whereas animal miRNAs end with free 2′, 3′ hydroxyl groups (80). Third, most plant miRNAs target mRNAs with near-perfect complementarity thereby inducing mRNA cleavage (81). Most animal miRNA targets have less complementarity thereby inducing translational suppression and sometimes degradation. Furthermore, there are examples of plant miRNAs that can direct methylation and transcriptional gene silencing (82), but to date, there are no examples of encoded animal miRNAs that do this.
Small interfering RNAs are processed from long double-stranded RNAs (dsRNAs) by Dicer (42). The resulting 21 nt long duplexes then enter the miRNA pathway and can induce mRNA cleavage, degradation, and translational suppression (83). Exogenous siRNAs have become a popular tool for gene knockdown, as researchers can design siRNAs for specific and effective knockdown of target genes (84,85). In mammals, long dsRNAs induce a strong interferon response, which usually leads to cell-death, but exogenous siRNAs can be introduced as 21 nt duplexes that mimic the Dicer product or as short (<30 nt) Dicer substrates (86–88). Exogenous siRNAs will usually only induce transient knockdown, but to create stable knockdown, one can instead express siRNAs as miRNA-mimicking hairpins (89–91).
Current literature describes few endogenous siRNAs, with some notable exceptions. Repeat-associated siRNAs (rasiRNAs) presumably arise from overlapping sense and antisense repeat-associated transcripts, regulate transposable elements pre and post transcription, and are involved in establishing and maintaining heterochromatin structure (92). Repeat-associated siRNAs may be crucial for protecting the germ line from transposable elements (93–95). Trans-acting siRNAs (tasiRNAs) arise from plant miRNAs that cleave nonprotein coding transcripts. The plant RdRp then uses the cleavage product as template for dsRNA generation, which leads to subsequent Dicer cleavage and siRNA generation (96). Small interfering RNAs can also arise from natural antisense transcripts (nat-siRNAs) (97).
Recently, a new class of small RNAs were identified in murine testes (95,98–101). These RNAs, which are located in strand-specific, nonoverlapping clusters throughout the genome, interact with the Piwi-subclass of Argonaute proteins—hence the name PIWI-interacting RNAs (piRNAs). The function of piRNAs is not clear, but as Piwi-family proteins associate with heterochromatic proteins and piRNA clusters are conserved in location and structure but not sequence, one could speculate that the piRNAs regulate or silence their own genomic regions.
MicroRNAs' RELATION TO EPIGENETICS
In the past few years, miRNAs have been established as enormously important mediators of gene regulation. Endogenous miRNAs are important in developmental processes, including differentiation, proliferation, and apoptosis (102). While classical epigenetic mechanisms, such as histone modification and DNA methylation, regulate expression at the transcriptional level, miRNAs putatively function mainly at the posttranscriptional level.
Link to classical mechanisms.
We have described the classical epigenetic mechanisms, namely DNA methylation and histone modification, as distinct mechanisms, but there is overwhelming evidence that this is not the case. Epigenetics mechanisms appear to be interconnected on multiple levels, and reports have shown three possible models [evidence summarized and models depicted in (7)]. The models propose that DNA methylation may direct histone methylation or vice versa—alternatively that chromatin remodeling drives DNA methylation. Of course, since the mechanisms appear to be interconnected, both models may be correct.
Whether miRNA regulation is an epigenetic mechanism in its own right is unclear, but several papers have described how miRNA expression is tissue-specific during development, which may implicate that miRNAs are crucial to establish and maintain cell type and tissue identity (60,103). Over-expression of tissue-specific miRNAs has also confirmed a single miRNA's potential to change the gene expression profile of a cell (71). Even though there have been speculations that animal miRNAs may be involved in the classical epigenetic mechanisms, as some plant miRNAs can direct methylation, the literature currently does not contain any evidence for direct endogenous miRNA involvement in transcriptional gene silencing. It has, however, been shown that RNA interference seems to be capable of inducing transcriptional gene silencing in cultured cells. In plants, small interfering RNAs (siRNA) are able to induce DNA methylation, which in turn gives transcriptional gene silencing (104,105). Whether this translates to animals has been and still is debated, and while early reports indicated that DNA methylation was involved in transcriptional gene silencing in mammalian cells (106), more recent evidence suggest that siRNA-induced transcriptional gene silencing is independent of DNA methylation (107).
A link between siRNA-induced transcriptional gene silencing and histone modifications has, however, been established. For example, Kim et al. (108) showed that Ago1 is required for histone H3 Lys9 dimethylation and transcriptional gene silencing. Furthermore, Janowsky et al. (109) found that Ago2 is also involved in addition to Ago1 (Fig. 2). The details on how the RNAi apparatus is involved in transcriptional gene silencing and the differences between organisms remain to be seen. Also, it will be interesting to see whether endogenous miRNAs or other RNAs are involved in transcriptional silencing in vivo or if the examples shown so far pertains strictly to artificial situations in vitro.

Proposed mechanism for small RNA triggered transcriptional gene silencing in mammals. SiRNAs complex with one or more of the Argonaute proteins, which direct the siRNA to a promoter region. There is evidence that promoter region transcripts may be required for this targeting, allowing sequence specific triggering of histone methylation and polycomb protein based remodeling of the chromatin, ultimately resulting in a heterochromatic status. The possible recruitment of DNA methyl transferases may follow the histone modifications.
Differences between organisms.
As mentioned previously, there are important differences in how miRNAs function in various species. In C. elegans, the effect of a miRNA can be maintained for a long time after the original stimuli for its expression has vanished. Single-stranded RNA that is complementary to the mRNA can serve as a primer for an RNA-directed RNA polymerase that can make long double-stranded RNA that can give rise to a wide range of siRNA species (110). Thus, silencing can be maintained, amplified, and even carried over to other genes without de novo expression of additional RNAs. Plants are also able to maintain silencing much the same way as worms do (111). Mammals, however, seem to lack an RNA-dependent RNA polymerase and are therefore unable to amplify and maintain RNA-mediated silencing without continuous expression of the RNA that mediates the effect. Consequently, siRNA-mediated RNAi is transient and the effect will usually last only through a few cell cycles without continuous expression of the RNA that mediates the effect (112).
In addition to the ability to maintain silencing, worms also inherit the effect from parent to offspring (113). Females that receive double-stranded RNA can transfer the effect to their offspring, and males can also transfer it to the subsequent generation. Note that this can happen even in the absence of the endogenous gene that is targeted, which means that RNAi can be inherited epigenetically in worms. Again, this has not been demonstrated to happen in mammals, which perhaps reflects the differences between RNA-mediated silencing in the various organisms.
CLINICAL RELEVANCE OF miRNA
Like epigenetic mechanisms, which have been shown to play a role in disorders ranging from various forms of cancer to syndromes involving behavioral disabilities, chromosome instability, organ overgrowth, and anemia (114), miRNAs appear to be important in the onset and development of several diseases. In humans, miRNAs' involvement in cancer has spurred a lot of interest (115). Curiously, many miRNAs are located at or close to genomic sites that are commonly deleted or amplified in various cancers (116). Also, expression of miRNAs is frequently decreased or increased in cancerous tissues. For example, miR-15 and miR-16 are often deleted or otherwise down-regulated in chronic lymphocytic leukemia (117), whereas miR-155 expression levels are elevated in human B-cell lymphomas (118). The interactions are likely context specific, and to illustrate the difficulties involved when assigning a cancer-related function to a miRNA, the mir-17-92 polycistron can function both as an oncogene and as a tumor suppressor (119,120). In addition to various cancers, miRNAs have also been implicated in genetic disorders, such as DiGeorge syndrome, and likely play a role in virus infection and defense.
Short hairpin RNA (shRNA) and small interfering RNA (siRNA)—the molecules that trigger stable and transient RNAi—are similar to the intermediates of miRNA biogenesis before and after Dicer processing. Consequently, an understanding of miRNA transcription, biogenesis, and function is important for therapeutic RNAi initiatives (121). Several side effects are known for RNAi. First, immunostimulatory effects, such as the interferon response, may be triggered by both shRNAs and siRNAs (122–124). Second, down-regulation of genes other than the intended target, so called off-target effects, are probably due to miRNA-like targeting and may cause widespread phenotypic effects (125–129). Third, since RNAi uses the same cellular factors as miRNAs, shRNAs and siRNAs may compete for and saturate critical enzymes and protein complexes needed for biogenesis and targeting. As previously mentioned, Exportin-5 may be rate limiting for miRNA biogenesis (40,41), which may explain why siRNAs appear not to saturate the pathway in vivo (130,131). Note that all of these problems can be mitigated by working at the lowest possible concentration, which means that more research into the miRNA pathway is needed to understand the consequences of therapeutic intervention using miRNA-like molecules; see (1,132) for reviews.
In addition to the therapeutic potential, miRNAs contain a lot of information about the regulatory state of the cells. In attempts to classify various forms of cancer with microarray expression profiles, miRNAs have been shown to harbor more information about disease states than do mRNA profiles (133). MicroRNA expression profiles therefore have the potential to become important diagnostic tools.
OUTLOOK
An impressive number of papers have been published on miRNAs since the discovery of these regulatory RNAs in 2001. But as the importance of miRNAs has grown, the number of scientific challenges has increased. Perhaps the most important task is to establish reliable models for how miRNAs target endogenous mRNAs. While a perfect model is unlikely due to the complexity of the problem, even significantly improved models will have a daunting effect on miRNA research. Coupled with data on miRNA expression in various tissues, reliable target prediction may allow simulations that may reveal important regulatory networks. Studies of how cells would react to aberrant miRNA expression may even become feasible if the target prediction methods improve sufficiently.
RNAi appears to be interconnected with DNA methylation and histone modifications. In some species, the link between miRNAs and epigenetics is strong. While the potential may be there, as shown in vitro by transfection of synthetic siRNAs, it remains to be seen whether these effects take place naturally. It will be interesting to see whether endogenous miRNAs or other types of nonprotein coding RNAs can be linked to epigenetic mechanisms in vivo. Also, an important question going forward is how miRNAs are involved in the establishment and maintenance of tissue-specific expression profiles. When the details on this become clearer, it is likely to reveal additional links to epigenetic mechanisms if they exist.
Finally, it is necessary to continue to research other nonprotein coding RNAs. The recent discovery of piRNAs—an abundant class of RNAs that were discovered in murine testes—shows that although miRNAs constitute an important part of the puzzle, we should be open to the possibility that additional classes of regulatory RNA may exist. Insight into the function of these additional classes of regulatory RNA and how they may involve all or some of the proteins in the miRNA pathway will be important to unravel all aspects of gene regulation. Naturally, all classes of regulatory RNA may be important players in what constitutes the epigenome.
References
Hannon GJ, Rossi JJ 2004 Unlocking the potential of the human genome with RNA interference. Nature 431: 371–378
Lagos-Quintana M, Rauhut R, Lendeckel W, Tuschl T 2001 Identification of novel genes coding for small expressed RNAs. Science 294: 853–858
Lau NC, Lim LP, Weinstein EG, Bartel DP 2001 An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science 294: 858–862
Lee RC, Ambros V 2001 An extensive class of small RNAs in Caenorhabditis elegans. Science 294: 862–864
Bird A 2002 DNA methylation patterns and epigenetic memory. Genes Dev 16: 6–21
Jenuwein T, Allis CD 2001 Translating the histone code. Science 293: 1074–1080
Li E 2002 Chromatin modification and epigenetic reprogramming in mammalian development. Nat Rev Genet 3: 662–673
Okano M, Bell DW, Haber DA, Li E 1999 DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 99: 247–257
Bestor TH 1992 Activation of mammalian DNA methyltransferase by cleavage of a Zn binding regulatory domain. EMBO J 11: 2611–2617
Goll MG, Kirpekar F, Maggert KA, Yoder JA, Hsieh CL, Zhang X, Golic KG, Jacobsen SE, Bestor TH 2006 Methylation of tRNAAsp by the DNA methyltransferase homolog Dnmt2. Science 311: 395–398
Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ 1997 Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 389: 251–260
Rassoulzadegan M, Grandjean V, Gounon P, Vincent S, Gillot I, Cuzin F 2006 RNA-mediated non-mendelian inheritance of an epigenetic change in the mouse. Nature 441: 469–474
Lee RC, Feinbaum RL, Ambros V 1993 The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 75: 843–854
Olsen PH, Ambros V 1999 The lin-4 regulatory RNA controls developmental timing in Caenorhabditis elegans by blocking LIN-14 protein synthesis after the initiation of translation. Dev Biol 216: 671–680
Reinhart BJ, Slack FJ, Basson M, Pasquinelli AE, Bettinger JC, Rougvie AE, Horvitz HR, Ruvkun G 2000 The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature 403: 901–906
Wightman B, Ha I, Ruvkun G 1993 Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell 75: 855–862
Griffiths-Jones S, Grocock RJ, van Dongen S, Bateman A, Enright AJ 2006 miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res 34: D140–D144
Bentwich I, Avniel A, Karov Y, Aharonov R, Gilad S, Barad O, Barzilai A, Einat P, Einav U, Meiri E, Sharon E, Spector Y, Bentwich Z 2005 Identification of hundreds of conserved and nonconserved human microRNAs. Nat Genet 37: 766–770
Berezikov E, van Tetering G, Verheul M, van de Belt J, van Laake L, Vos J, Verloop R, van de Wetering M, Guryev V, Takada S, van Zonneveld AJ, Mano H, Plasterk R, Cuppen E 2006 Many novel mammalian microRNA candidates identified by extensive cloning and RAKE analysis. Genome Res 16: 1289–1298
Berezikov E, Guryev V, van de Belt J, Wienholds E, Plasterk RH, Cuppen E 2005 Phylogenetic shadowing and computational identification of human microRNA genes. Cell 120: 21–24
Ason B, Darnell DK, Wittbrodt B, Berezikov E, Kloosterman WP, Wittbrodt J, Antin PB, Plasterk RH 2006 Differences in vertebrate microRNA expression. Proc Natl Acad Sci U S A 103: 14385–14389
Cai X, Hagedorn CH, Cullen BR 2004 Human microRNAs are processed from capped, polyadenylated transcripts that can also function as mRNAs. RNA 10: 1957–1966
Lee Y, Kim M, Han J, Yeom KH, Lee S, Baek SH, Kim VN 2004 MicroRNA genes are transcribed by RNA polymerase II. EMBO J 23: 4051–4060
Lagos-Quintana M, Rauhut R, Meyer J, Borkhardt A, Tuschl T 2003 New microRNAs from mouse and human. RNA 9: 175–179
Karolchik D, Baertsch R, Diekhans M, Furey TS, Hinrichs A, Lu YT, Roskin KM, Schwartz M, Sugnet CW, Thomas DJ, Weber RJ, Haussler D, Kent WJ 2003 The UCSC Genome Browser Database. Nucleic Acids Res 31: 51–54
Smalheiser NR, Torvik VI 2005 Mammalian microRNAs derived from genomic repeats. Trends Genet 21: 322–326
Altuvia Y, Landgraf P, Lithwick G, Elefant N, Pfeffer S, Aravin A, Brownstein MJ, Tuschl T, Margalit H 2005 Clustering and conservation patterns of human microRNAs. Nucleic Acids Res 33: 2697–2706
Baskerville S, Bartel DP 2005 Microarray profiling of microRNAs reveals frequent coexpression with neighboring miRNAs and host genes. RNA 11: 241–247
Denli AM, Tops BB, Plasterk RH, Ketting RF, Hannon GJ 2004 Processing of primary microRNAs by the Microprocessor complex. Nature 432: 231–235
Gregory RI, Yan KP, Amuthan G, Chendrimada T, Doratotaj B, Cooch N, Shiekhattar R 2004 The Microprocessor complex mediates the genesis of microRNAs. Nature 432: 235–240
Han J, Lee Y, Yeom KH, Kim YK, Jin H, Kim VN 2004 The Drosha-DGCR8 complex in primary microRNA processing. Genes Dev 18: 3016–3027
Lee Y, Ahn C, Han J, Choi H, Kim J, Yim J, Lee J, Provost P, Radmark O, Kim S, Kim VN 2003 The nuclear RNase III Drosha initiates microRNA processing. Nature 425: 415–419
Han J, Lee Y, Yeom KH, Nam JW, Heo I, Rhee JK, Sohn SY, Cho Y, Zhang BT, Kim VN 2006 Molecular basis for the recognition of primary microRNAs by the Drosha-DGCR8 complex. Cell 125: 887–901
Saetrom P, Snove O, Nedland M, Grunfeld TB, Lin Y, Bass MB, Canon JR 2006 Conserved MicroRNA characteristics in mammals. Oligonucleotides 16: 115–144
Zeng Y, Yi R, Cullen BR 2005 Recognition and cleavage of primary microRNA precursors by the nuclear processing enzyme Drosha. EMBO J 24: 138–148
Bohnsack MT, Czaplinski K, Gorlich D 2004 Exportin 5 is a RanGTP-dependent dsRNA-binding protein that mediates nuclear export of pre-miRNAs. RNA 10: 185–191
Lund E, Guttinger S, Calado A, Dahlberg JE, Kutay U 2004 Nuclear export of microRNA precursors. Science 303: 95–98
Yi R, Qin Y, Macara IG, Cullen BR 2003 Exportin-5 mediates the nuclear export of pre-microRNAs and short hairpin RNAs. Genes Dev 17: 3011–3016
Zeng Y, Cullen BR 2004 Structural requirements for pre-microRNA binding and nuclear export by Exportin 5. Nucleic Acids Res 32: 4776–4785
Grimm D, Streetz KL, Jopling CL, Storm TA, Pandey K, Davis CR, Marion P, Salazar F, Kay MA 2006 Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways. Nature 441: 537–541
Yi R, Doehle BP, Qin Y, Macara IG, Cullen BR 2005 Overexpression of exportin 5 enhances RNA interference mediated by short hairpin RNAs and microRNAs. RNA 11: 220–226
Bernstein E, Caudy AA, Hammond SM, Hannon GJ 2001 Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature 409: 363–366
Hutvagner G, McLachlan J, Pasquinelli AE, Balint E, Tuschl T, Zamore PD 2001 A cellular function for the RNA-interference enzyme Dicer in the maturation of the let-7 small temporal RNA. Science 293: 834–838
Vermeulen A, Behlen L, Reynolds A, Wolfson A, Marshall WS, Karpilow J, Khvorova A 2005 The contributions of dsRNA structure to Dicer specificity and efficiency. RNA 11: 674–682
Chendrimada TP, Gregory RI, Kumaraswamy E, Norman J, Cooch N, Nishikura K, Shiekhattar R 2005 TRBP recruits the Dicer complex to Ago2 for microRNA processing and gene silencing. Nature 436: 740–744
Gregory RI, Chendrimada TP, Cooch N, Shiekhattar R 2005 Human RISC couples microRNA biogenesis and posttranscriptional gene silencing. Cell 123: 631–640
Matranga C, Tomari Y, Shin C, Bartel DP, Zamore PD 2005 Passenger-strand cleavage facilitates assembly of siRNA into Ago2-containing RNAi enzyme complexes. Cell 123: 607–620
Rand TA, Petersen S, Du F, Wang X 2005 Argonaute2 cleaves the anti-guide strand of siRNA during RISC activation. Cell 123: 621–629
Tomari Y, Matranga C, Haley B, Martinez N, Zamore PD 2004 A protein sensor for siRNA asymmetry. Science 306: 1377–1380
Khvorova A, Reynolds A, Jayasena SD 2003 Functional siRNAs and miRNAs exhibit strand bias. Cell 115: 209–216
Schwarz DS, Hutvagner G, Du T, Xu Z, Aronin N, Zamore PD 2003 Asymmetry in the assembly of the RNAi enzyme complex. Cell 115: 199–208
Obernosterer G, Leuschner PJ, Alenius M, Martinez J 2006 Post-transcriptional regulation of microRNA expression. RNA 12: 1161–1167
Thomson JM, Newman M, Parker JS, Morin-Kensicki EM, Wright T, Hammond SM 2006 Extensive post-transcriptional regulation of microRNAs and its implications for cancer. Genes Dev 20: 2202–2207
Ma JB, Yuan YR, Meister G, Pei Y, Tuschl T, Patel DJ 2005 Structural basis for 5′-end-specific recognition of guide RNA by the A. fulgidus Piwi protein. Nature 434: 666–670
Parker JS, Roe SM, Barford D 2005 Structural insights into mRNA recognition from a PIWI domain-siRNA guide complex. Nature 434: 663–666
Rivas FV, Tolia NH, Song JJ, Aragon JP, Liu J, Hannon GJ, Joshua-Tor L 2005 Purified Argonaute2 and an siRNA form recombinant human RISC. Nat Struct Mol Biol 12: 340–349
Yekta S, Shih IH, Bartel DP 2004 MicroRNA-directed cleavage of HOXB8 mRNA. Science 304: 594–596
Zamore PD, Tuschl T, Sharp PA, Bartel DP 2000 RNAi: double-stranded RNA directs the ATP-dependent cleavage of mRNA at 21 to 23 nucleotide intervals. Cell 101: 25–33
Bagga S, Bracht J, Hunter S, Massirer K, Holtz J, Eachus R, Pasquinelli AE 2005 Regulation by let-7 and lin-4 miRNAs results in target mRNA degradation. Cell 122: 553–563
Giraldez AJ, Cinalli RM, Glasner ME, Enright AJ, Thomson JM, Baskerville S, Hammond SM, Bartel DP, Schier AF 2005 MicroRNAs regulate brain morphogenesis in zebrafish. Science 308: 833–838
Wu L, Fan J, Belasco JG 2006 MicroRNAs direct rapid deadenylation of mRNA. Proc Natl Acad Sci U S A 103: 4034–4039
Amarzguioui M, Holen T, Babaie E, Prydz H 2003 Tolerance for mutations and chemical modifications in a siRNA. Nucleic Acids Res 31: 589–595
Du Q, Thonberg H, Wang J, Wahlestedt C, Liang Z 2005 A systematic analysis of the silencing effects of an active siRNA at all single-nucleotide mismatched target sites. Nucleic Acids Res 33: 1671–1677
Elbashir SM, Martinez J, Patkaniowska A, Lendeckel W, Tuschl T 2001 Functional anatomy of siRNAs for mediating efficient RNAi in Drosophila melanogaster embryo lysate. EMBO J 20: 6877–6888
Schwarz DS, Ding H, Kennington L, Moore JT, Schelter J, Burchard J, Linsley PS, Aronin N, Xu Z, Zamore PD 2006 Designing siRNA that distinguish between genes that differ by a single nucleotide. PLoS Genet 2: e140
Brennecke J, Stark A, Russell RB, Cohen SM 2005 Principles of microRNA-target recognition. PLoS Biol 3: e85.
Krek A, Grun D, Poy MN, Wolf R, Rosenberg L, Epstein EJ, MacMenamin P, da Piedade I, Gunsalus KC, Stoffel M, Rajewsky N 2005 Combinatorial microRNA target predictions. Nat Genet 37: 495–500
Lewis BP, Burge CB, Bartel DP 2005 Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120: 15–20
Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB 2003 Prediction of mammalian microRNA targets. Cell 115: 787–798
Xie X, Lu J, Kulbokas EJ, Golub TR, Mootha V, Lindblad-Toh K, Lander ES, Kellis M 2005 Systematic discovery of regulatory motifs in human promoters and 3′ UTRs by comparison of several mammals. Nature 434: 338–345
Lim LP, Lau NC, Garrett-Engele P, Grimson A, Schelter JM, Castle J, Bartel DP, Linsley PS, Johnson JM 2005 Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature 433: 769–773
Vella MC, Choi EY, Lin SY, Reinert K, Slack FJ 2004 The C. elegans microRNA let-7 binds to imperfect let-7 complementary sites from the lin-41 3′UTR. Genes Dev 18: 132–137
Vella MC, Reinert K, Slack FJ 2004 Architecture of a validated microRNA:target interaction. Chem Biol 11: 1619–1623
Didiano D, Hobert O 2006 Perfect seed pairing is not a generally reliable predictor for miRNA-target interactions. Nat Struct Mol Biol 13: 849–851
Aravin A, Tuschl T 2005 Identification and characterization of small RNAs involved in RNA silencing. FEBS Lett 579: 5830–5840
Bartel DP 2004 MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116: 281–297
Du T, Zamore PD 2005 microPrimer: the biogenesis and function of microRNA. Development 132: 4645–4652
Mallory AC, Vaucheret H 2006 Functions of microRNAs and related small RNAs in plants. Nat Genet 38: S31–S36
Kurihara Y, Watanabe Y 2004 Arabidopsis micro-RNA biogenesis through Dicer-like 1 protein functions. Proc Natl Acad Sci U S A 101: 12753–12758
Yu B, Yang Z, Li J, Minakhina S, Yang M, Padgett RW, Steward R, Chen X 2005 Methylation as a crucial step in plant microRNA biogenesis. Science 307: 932–935
Rhoades MW, Reinhart BJ, Lim LP, Burge CB, Bartel B, Bartel DP 2002 Prediction of plant microRNA targets. Cell 110: 513–520
Bao N, Lye KW, Barton MK 2004 MicroRNA binding sites in Arabidopsis class III HD-ZIP mRNAs are required for methylation of the template chromosome. Dev Cell 7: 653–662
Doench JG, Petersen CP, Sharp PA 2003 siRNAs can function as miRNAs. Genes Dev 17: 438–442
Pei Y, Tuschl T 2006 On the art of identifying effective and specific siRNAs. Nat Methods 3: 670–676
Saetrom P, Snove O Jr 2004 A comparison of siRNA efficacy predictors. Biochem Biophys Res Commun 321: 247–253
Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T 2001 Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 411: 494–498
Kim DH, Behlke MA, Rose SD, Chang MS, Choi S, Rossi JJ 2005 Synthetic dsRNA Dicer substrates enhance RNAi potency and efficacy. Nat Biotechnol 23: 222–226
Siolas D, Lerner C, Burchard J, Ge W, Linsley PS, Paddison PJ, Hannon GJ, Cleary MA 2005 Synthetic shRNAs as potent RNAi triggers. Nat Biotechnol 23: 227–231
Chang K, Elledge SJ, Hannon GJ 2006 Lessons from nature: microRNA-based shRNA libraries. Nat Methods 3: 707–714
McManus MT, Petersen CP, Haines BB, Chen J, Sharp PA 2002 Gene silencing using micro-RNA designed hairpins. RNA 8: 842–850
Paddison PJ, Caudy AA, Bernstein E, Hannon GJ, Conklin DS 2002 Short hairpin RNAs (shRNAs) induce sequence-specific silencing in mammalian cells. Genes Dev 16: 948–958
Lippman Z, Martienssen R 2004 The role of RNA interference in heterochromatic silencing. Nature 431: 364–370
Saito K, Nishida KM, Mori T, Kawamura Y, Miyoshi K, Nagami T, Siomi H, Siomi MC 2006 Specific association of Piwi with rasiRNAs derived from retrotransposon and heterochromatic regions in the Drosophila genome. Genes Dev 20: 2214–2222
Vagin VV, Sigova A, Li C, Seitz H, Gvozdev V, Zamore PD 2006 A distinct small RNA pathway silences selfish genetic elements in the germline. Science 313: 320–324
Watanabe T, Takeda A, Tsukiyama T, Mise K, Okuno T, Sasaki H, Minami N, Imai H 2006 Identification and characterization of two novel classes of small RNAs in the mouse germline: retrotransposon-derived siRNAs in oocytes and germline small RNAs in testes. Genes Dev 20: 1732–1743
Allen E, Xie Z, Gustafson AM, Carrington JC 2005 microRNA-directed phasing during trans-acting siRNA biogenesis in plants. Cell 121: 207–221
Borsani O, Zhu J, Verslues PE, Sunkar R, Zhu JK 2005 Endogenous siRNAs derived from a pair of natural cis-antisense transcripts regulate salt tolerance in Arabidopsis. Cell 123: 1279–1291
Aravin A, Gaidatzis D, Pfeffer S, Lagos-Quintana M, Landgraf P, Iovino N, Morris P, Brownstein MJ, Kuramochi-Miyagawa S, Nakano T, Chien M, Russo JJ, Ju J, Sheridan R, Sander C, Zavolan M, Tuschl T 2006 A novel class of small RNAs bind to MILI protein in mouse testes. Nature 442: 203–207
Girard A, Sachidanandam R, Hannon GJ, Carmell MA 2006 A germline-specific class of small RNAs binds mammalian Piwi proteins. Nature 442: 199–202
Grivna ST, Beyret E, Wang Z, Lin H 2006 A novel class of small RNAs in mouse spermatogenic cells. Genes Dev 20: 1709–1714
Lau NC, Seto AG, Kim J, Kuramochi-Miyagawa S, Nakano T, Bartel DP, Kingston RE 2006 Characterization of the piRNA complex from rat testes. Science 313: 363–367
Ambros V 2004 The functions of animal microRNAs. Nature 431: 350–355
Wienholds E, Kloosterman WP, Miska E, Alvarez-Saavedra E, Berezikov E, de Bruijn E, Horvitz HR, Kauppinen S, Plasterk RH 2005 MicroRNA expression in zebrafish embryonic development. Science 309: 310–311
Pelissier T, Thalmeir S, Kempe D, Sanger HL, Wassenegger M 1999 Heavy de novo methylation at symmetrical and non-symmetrical sites is a hallmark of RNA-directed DNA methylation. Nucleic Acids Res 27: 1625–1634
Wassenegger M, Heimes S, Riedel L, Sanger HL 1994 RNA-directed de novo methylation of genomic sequences in plants. Cell 76: 567–576
Morris KV, Chan SW, Jacobsen SE, Looney DJ 2004 Small interfering RNA-induced transcriptional gene silencing in human cells. Science 305: 1289–1292
Ting AH, Schuebel KE, Herman JG, Baylin SB 2005 Short double-stranded RNA induces transcriptional gene silencing in human cancer cells in the absence of DNA methylation. Nat Genet 37: 906–910
Kim DH, Villeneuve LM, Morris KV, Rossi JJ 2006 Argonaute-1 directs siRNA-mediated transcriptional gene silencing in human cells. Nat Struct Mol Biol 13: 793–797
Janowski BA, Huffman KE, Schwartz JC, Ram R, Nordsell R, Shames DS, Minna JD, Corey DR 2006 Involvement of AGO1 and AGO2 in mammalian transcriptional silencing. Nat Struct Mol Biol 13: 787–792
Sijen T, Fleenor J, Simmer F, Thijssen KL, Parrish S, Timmons L, Plasterk RH, Fire A 2001 On the role of RNA amplification in dsRNA-triggered gene silencing. Cell 107: 465–476
Dalmay T, Hamilton A, Rudd S, Angell S, Baulcombe DC 2000 An RNA-dependent RNA polymerase gene in Arabidopsis is required for posttranscriptional gene silencing mediated by a transgene but not by a virus. Cell 101: 543–553
Elbashir SM, Harborth J, Weber K, Tuschl T 2002 Analysis of gene function in somatic mammalian cells using small interfering RNAs. Methods 26: 199–213
Grishok A, Tabara H, Mello CC 2000 Genetic requirements for inheritance of RNAi in C. elegans. Science 287: 2494–2497
Egger G, Liang G, Aparicio A, Jones PA 2004 Epigenetics in human disease and prospects for epigenetic therapy. Nature 429: 457–463
Esquela-Kerscher A, Slack FJ 2006 Oncomirs - microRNAs with a role in cancer. Nat Rev Cancer 6: 259–269
Calin GA, Sevignani C, Dumitru CD, Hyslop T, Noch E, Yendamuri S, Shimizu M, Rattan S, Bullrich F, Negrini M, Croce CM 2004 Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc Natl Acad Sci U S A 101: 2999–3004
Calin GA, Dumitru CD, Shimizu M, Bichi R, Zupo S, Noch E, Aldler H, Rattan S, Keating M, Rai K, Rassenti L, Kipps T, Negrini M, Bullrich F, Croce CM 2002 Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci U S A 99: 15524–15529
Eis PS, Tam W, Sun L, Chadburn A, Li Z, Gomez MF, Lund E, Dahlberg JE 2005 Accumulation of miR-155 and BIC RNA in human B cell lymphomas. Proc Natl Acad Sci U S A 102: 3627–3632
He L, Thomson JM, Hemann MT, Hernando-Monge E, Mu D, Goodson S, Powers S, Cordon-Cardo C, Lowe SW, Hannon GJ, Hammond SM 2005 A microRNA polycistron as a potential human oncogene. Nature 435: 828–833
O'Donnell KA, Wentzel EA, Zeller KI, Dang CV, Mendell JT 2005 c-Myc-regulated microRNAs modulate E2F1 expression. Nature 435: 839–843
Behlke MA 2006 Progress towards in vivo use of siRNAs. Mol Ther 13: 644–670
Bridge AJ, Pebernard S, Ducraux A, Nicoulaz AL, Iggo R 2003 Induction of an interferon response by RNAi vectors in mammalian cells. Nat Genet 34: 263–264
Marques JT, Williams BR 2005 Activation of the mammalian immune system by siRNAs. Nat Biotechnol 23: 1399–1405
Sledz CA, Holko M, de Veer MJ, Silverman RH, Williams BR 2003 Activation of the interferon system by short-interfering RNAs. Nat Cell Biol 5: 834–839
Birmingham A, Anderson EM, Reynolds A, Ilsley-Tyree D, Leake D, Fedorov Y, Baskerville S, Maksimova E, Robinson K, Karpilow J, Marshall WS, Khvorova A 2006 3′ UTR seed matches, but not overall identity, are associated with RNAi off-targets. Nat Methods 3: 199–204
Fedorov Y, Anderson EM, Birmingham A, Reynolds A, Karpilow J, Robinson K, Leake D, Marshall WS, Khvorova A 2006 Off-target effects by siRNA can induce toxic phenotype. RNA 12: 1188–1196
Jackson AL, Bartz SR, Schelter J, Kobayashi SV, Burchard J, Mao M, Li B, Cavet G, Linsley PS 2003 Expression profiling reveals off-target gene regulation by RNAi. Nat Biotechnol 21: 635–637
Jackson AL, Burchard J, Schelter J, Chau BN, Cleary M, Lim L, Linsley PS 2006 Widespread siRNA “off-target” transcript silencing mediated by seed region sequence complementarity. RNA 12: 1179–1187
Lin X, Ruan X, Anderson MG, McDowell JA, Kroeger PE, Fesik SW, Shen Y 2005 siRNA-mediated off-target gene silencing triggered by a 7 nt complementation. Nucleic Acids Res 33: 4527–4535
Soutschek J, Akinc A, Bramlage B, Charisse K, Constien R, Donoghue M, Elbashir S, Geick A, Hadwiger P, Harborth J, John M, Kesavan V, Lavine G, Pandey RK, Racie T, Rajeev KG, Rohl I, Toudjarska I, Wang G, Wuschko S, Bumcrot D, Koteliansky V, Limmer S, Manoharan M, Vornlocher HP 2004 Therapeutic silencing of an endogenous gene by systemic administration of modified siRNAs. Nature 432: 173–178
Zimmermann TS, Lee AC, Akinc A, Bramlage B, Bumcrot D, Fedoruk MN, Harborth J, Heyes JA, Jeffs LB, John M, Judge AD, Lam K, McClintock K, Nechev LV, Palmer LR, Racie T, Rohl I, Seiffert S, Shanmugam S, Sood V, Soutschek J, Toudjarska I, Wheat AJ, Yaworski E, Zedalis W, Koteliansky V, Manoharan M, Vornlocher HP, MacLachlan I 2006 RNAi-mediated gene silencing in non-human primates. Nature 441: 111–114
Snove O Jr, Rossi JJ 2006 Toxicity in mice expressing short hairpin RNAs gives new insight into RNAi. Genome Biol 7: 231.
Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D, Sweet-Cordero A, Ebert BL, Mak RH, Ferrando AA, Downing JR, Jacks T, Horvitz HR, Golub TR 2005 MicroRNA expression profiles classify human cancers. Nature 435: 834–838
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Sætrom, P., Snøve, O. & Rossi, J. Epigenetics and MicroRNAs. Pediatr Res 61, 17–23 (2007). https://doi.org/10.1203/pdr.0b013e318045760e
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/pdr.0b013e318045760e
This article is cited by
-
Association of miRNA − 320 expression level and its target gene endothelin-1 with the susceptibility and clinical features of polycystic ovary syndrome
Journal of Ovarian Research (2019)
-
Extracellular microRNAs in follicular fluid and their potential association with oocyte fertilization and embryo quality: an exploratory study
Journal of Assisted Reproduction and Genetics (2017)
-
Silencing of microRNA-122 enhances interferon-α signaling in the liver through regulating SOCS3 promoter methylation
Scientific Reports (2012)
-
Neuroscience of alcoholism: molecular and cellular mechanisms
Cellular and Molecular Life Sciences (2010)
