
Abstract
Low birth weight (LBW) infants with reduced nephron numbers have significantly increased risk for hypertension later in life, which is a devastating health problem. The risk from a reduction in nephron number alone is not clear. Recently, using conditional knock-out approach, we have developed a mutant mouse with reduced nephron number in utero and no change in birth weight, by deleting fibroblast growth factor receptor 2 (fgfr2) in the ureteric bud. Our purpose was to investigate the role of in utero reduced nephron number alone in absence of LBW as a risk for developing hypertension in adulthood. Using tail cuff blood pressure measurements we observed significant increases in systolic blood pressure in one year old mutant mice versus controls. We also detected cardiac end-organ injury from hypertension as shown by significant increases in normalized heart weights, left ventricular (LV) wall thickness, and LV tissue area. Two-dimensional echocardiography revealed no changes in cardiac output and therefore significant increases in systemic vascular resistance in mutants versus controls. We also observed increases in serum blood urea nitrogen (BUN) levels and histologic evidence of glomerular and renal tubular injury in mutant mice versus controls. Thus, these studies suggest that our mutant mice may serve as a relevant model to study the link between reduction of nephron number in utero and the risk of hypertension and chronic renal failure in adulthood.
Similar content being viewed by others

Main
Hypertension is a chronic disease with an incidence of more than 65 million people in the US (1). According to recent estimates nearly one in three US adults have high blood pressure (1). This chronic and progressive disease is a leading cause of renal failure as well as increasing risks of stroke, heart disease and blindness.
Alarmingly, 7.8% of all infants born in the United States in 2002 had a LBW (<2500 g) (2); the majority of the LBW infants have low nephron numbers and an increased risk of hypertension in adulthood (3,4). Recently a clinical study has reported finding elevated systolic blood pressure in adult women born preterm (5). In addition, a meta-analysis that reviewed studies published between 1996 and 2000, concluded that birth weight was inversely related to systolic blood pressure (6). In 1988 Brenner et al. proposed that nephron number was inversely correlated to the risk of development of hypertension (7). Further evidence in support of this concept was recently documented in patients with a history of primary hypertension or LV hypertrophy who had significantly increased heart weights but fewer glomeruli per kidney than matched normotensive controls (8).
Most of the experimental models to study the link between hypertension and reduced number of nephrons or LBW have used either unilateral nephrectomy models (9,10) or models with intrauterine growth reduction (11–14). Although the collective data suggests a strong correlation with developing hypertension, the independent roles of LBW or reduced nephron numbers alone as important risk factors in causing hypertension later in life are not clarified. One previously published report using glial cell line-derived neurotrophic factor (GDNF) heterozygous mice suggested a link between in utero reduction of nephron number alone and increased risk of hypertension (15).
Recently, using a conditional knock out approach, we have developed a mutant mouse with reduced nephron number in utero and no change in birth weight, by deleting fgfr2 in the ureteric bud (16). The mice formed normal-appearing nephrons and appeared to have normal birth weights; however, we observed a 24% decrease in nephron number in mutant mice both in embryonic explants and in 2 mo old adult mice versus controls (14). This has given us a unique opportunity to study the role of in utero reduction of nephron number on progression of hypertension, changes in cardiac function, and changes in kidney function in the absence of LBW.
The purpose of our study was to further clarify the independent role of reduced nephron number on increased risk for hypertension in adulthood. In addition our study focused on an extensive investigation of cardiac profiling including echocardiography and cardiac remodeling in this model. Finally we examined the effects of renal function in mutant versus control mice
MATERIALS AND METHODS
Animals.
All animal handling protocols were approved by Institutional Laboratory Animal Care and Use Committee at our institution. The generation of the conditional fgfr2 knock out mice has been previously described (16). Briefly, mice with lox-p sites flanking critical regions of fgfr2 in the ureteric bud were bred with transgenic Hoxb7creEGFP (Hoxcre) mice to obtain subsequent Hoxcre ± mice with deletion of fgfr2 from ureteric bud (mutants) and Hoxcre−/− mice (controls). Mutant (n = 6) and littermate controls (n = 8) were housed in a 12-h light/dark facility with free access to food and water. All mice were fed Teklad Global 18% Protein Rodent Diet (Harlan Teklad, Madison, Wisconsin), which has 0.23% sodium content. The mice were weighed at birth and then at one year of age with the genotypes blinded to investigator. Genotyping for cre and fgfr2 alleles were by PCR as previously described (16). The mice were studied at approximately 1 y of age at which time the animals were anesthetized using 1% to 2% isoflurane, delivered by a small nose cone.
Blood pressure measurements.
Following anesthetic induction, a Harvard Tail BP monitor (Harvard Apparatus, Holliston, MA) was used to measure systolic blood pressures of both the mutant and control animals. All measurements were repeated three times at 10 min intervals and performed at the same time of the day.
Echocardiography and Doppler flow analysis.
Cardiac performance was assessed by echocardiography as previously described (17,18). Briefly, following anesthetic induction, mice were gently restrained in the left lateral decubitus position with elastic bands attached to a heated pad to maintain normothermia. The chest area was shaved and ultrasound coupling gel was liberally applied to the left chest wall. Two dimensional and M-mode echocardiographic images were recorded and analyzed by a Sonos 1000 echocardiograph and a 15 MHz pediatric ultrasonic probe (Hewlett-Packard Company, Andover, MA). Two-dimensional transverse LV imaging was used to position the probe just distal to the mitral valve leaflets and M-mode images were then captured. Three loops of M-mode data were captured from each animal at approximately 5 min intervals and stored on a digital disk until analyzed. Each of these captured image loops provided 7–12 heart cycles. Data were averaged from at least 5 cycles/loop. LV systolic (LVIDs) and diastolic (LVIDd) internal dimensions were measured according to the American Society for Echocardiography leading-edge technique by a blinded investigator. These parameters allowed the determination of LV fractional shortening (%FS), a measure of systolic function, by the equation: %FS = [(LVIDd-LVIDs)/ LVIDd]×100%.
Ascending aortic flow velocity was determined using the ultrasonic probe in continuous Doppler wave mode. The probe was maintained in the parasternal short axis orientation, but moved horizontally along the chest wall toward the suprasternal notch. While monitoring real time color flow, the probe was slowly angled toward the head of the animal (probe face pointing toward the heart). This probe position provided color-enhanced definition of blood flow around the aortic arch. The Doppler beam was centered on the ascending flow tract approximately 2 mm distal from the aortic valves and beat-to-beat cycles of aortic blood flow velocity were then recorded in three captured loops as described above. From these recordings, peak aortic flow velocity and velocity-time integral (VTI) was determined for at least 5 beats/loop for each animal. After sacrifice, aortic root cross-sectional area was measured, and cardiac output (CO) was calculated by the equation: CO = heart rate × VTI × aortic cross-sectional area.
Histochemistry and digital image analysis.
Animals were killed with 100 mg/kg, intra peritoneal, pentobarbital sodium (Abbott Laboratories, Chicago, IL). The apical portion of the heart was bisected just distal to the mitral valve and immersed in 10% formalin. Kidneys were weighed and then bisected through the hilum in the transverse plane. Following a 48-h fixation period in 10% buffered formalin, tissues were dehydrated and paraffin-embedded using standard procedures, as previously described (17). Serial 5-μm tissue sections were placed on microscope slides, dewaxed, and rehydrated for routine histochemical staining (hematoxylin & eosin) for morphologic studies. Photomicrographs were captured on a Polaroid DMC high-resolution digital camera, mounted on an Olympus research microscope (Pixera Inc., Houston, TX). The images were analyzed with Image Pro 5.0 image analysis software (Media Cybernetics, Silver Spring, MD). Hematoxylin and eosin stained slides were used to determine LV wall thickness (at the equatorial midline). Each heart cross-section was captured in its entirety at 2.5× magnification and internal and external edges were traced (automated edge detection, Image Pro 5.0) for the left ventricle to determine the LV wall thickness. For all parameters measured, intra- and inter observer variabilities were less than 5% (coefficients of variation for three daily measurements made by three different investigators evaluating three hearts).
Renal Profiling.
Before sacrifice, 24 h urine samples were obtained for measurement of urine chemistries and blood samples were collected by cardiac puncture, at the time of sacrifice, to measure serum chemistries (electrolytes, creatinine, BUN, potassium, bicarbonate calcium, osmolality). Serum and urine urea and creatinine levels were measured using a VITROS apparatus (Ortho-Clinical diagnostics, Rochester, NY) at the animal Morphology Core (Ohio State University, Columbus, OH). Serum and urine electrolytes were analyzed by a Hitachi 911 apparatus (Roche, Indianapolis, IN) at the animal Morphology Core. Heart tissue was weighed and rapidly isolated for histology and/or snap frozen for biochemical analysis.
Statistics.
All statistical evaluations were performed using SigmaStat statistical software (Jandel Scientific, San Rafael, CA). Statistical comparisons were determined using t test. In all tests p < 0.05 were considered significant.
RESULTS
Body weights in mutant versus control mice.
We detected no statistically significant changes in the body weights of the newborn mutant mice compared with controls (1.43 ± 0.09 versus 1.50 ± 0.07 g, p = 0.6). Similarly, we observed no statistically significant changes in the body weights of the adult (1-y-old) (34.4 ± 1.9 versus 35.6 ± 1.4 g, p = 0.42) mutant mice versus controls.
Blood pressure profiling and cardiac remodeling in adult mutant mice.
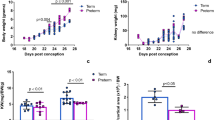
We detected striking increases in systolic blood pressures in adult mutant mice versus control littermates (133 ± 7 versus 113 ± 3 mm Hg, p = 0.001), as shown in Fig. 1. To determine whether the mutants developed end organ damage from increased blood pressures we examined the mutants for cardiac remodeling. Representative photomicrographs (2.5× magnification) of heart cross-sections through equatorial midline reveal significant increases in overall mutant left ventricle size as compared with controls (Fig. 2A,B). Quantitatively, heart weights normalized to body weights were significantly increased in mutants versus controls (0.004 ± 0.002 versus 0.0056 ± 0.004 g/g, p < 0.05) (Fig. 2C). In addition, overall LV area (area between outer and inner lines in Fig. 2A, 2B) was significantly increased in mutants versus controls (Fig. 2D). Finally average LV wall thickness calculated by digital image analysis software, which traced the outer and inner edges of the left ventricle (Fig. E2) was dramatically increased in mutant versus control mice (0.78 ± 0.09 versus 0.55 ± 0.058 mm, p < 0.05). Thus hypertension, as demonstrated by increased blood pressure and cardiac remodeling was clearly present in the mutant mice.

Mean systolic blood pressure measurements in mutant and control mice. Mutants (Mut) (n = 6) have significant increases in systolic BP compared with controls (Con) (n = 8). Data shown are mean and SEM. *Statistically significant from control, p < 0.05.

Cardiac remodeling in mutant mice. Representative photomicrographs of H&E stained transverse sections of hearts for 1-y-old controls (A) and mutant mice (B) (both 2.5 × magnification). Significant increases in heart weight (C), LV tissue area (D), and LV wall thickness (E) were observed in mutant (Mut) (n = 3) versus control (Con) (n = 4) mice. Outer and inner border of the control left ventricle are outlined and LV thickness is indicated by an arrow (A). Data shown in C-E are mean and SEM. *Statistically significant from control, p < 0.05. Scale bar for A, B = 0.5 mm.
Cardiac profiling.
Representative two-dimensional M-mode tracings of systole-diastole cycles from a control and mutant animal are shown in Fig. 3A (top panel). The waveforms from control and mutant animals were not different, consistent with no alterations in the fractional shortening (49.8 ± 2.2 versus 49.1 ± 2.47%, p = 0.84). Resting heart rate (440 ± 23.6 versus 500 ± 18.9 bpm, p = 0.07) was not different between the groups. Although there was a trend for decreased cardiac output (14.67 ± 2.02 versus 19.78 ± 1.5 mL/min, p = 0.06) and increased stroke volume (33.85 ± 4.67 versus 39.38 ± 2.08 μL, p = 0.26) in mutant mice versus controls, we observed no significant changes in either parameter. Systemic vascular resistance was dramatically increased in mutant mice versus controls (11.31 ± 3.1 versus 6.13 ± 0.38 mm Hg × min/mL, p < 0.05) (Fig. 3B). Thus there are no differences in cardiac output but significant increases in systemic vascular resistance in mutant mice versus controls.

Cardiac profiling in mutant and control mice. Representative two-dimensional M-mode tracings of systole-diastole cycles from a control (Con) (upper panel) and mutant (Mut) (lower panel) animal are shown (A). There was a significant two –fold increase in the systemic vascular resistance in the mutant (Mut) mice (n = 6) versus controls (Con) (n = 8) (B). Data shown in B are mean and SEM. *Statistically significant from control, p < 0.05.
Renal profiling.
We observed significant increases in serum BUN levels (37.2 ± 7.6 versus 22.8 ± 3.8 mg/dL, p < 0.05) in mutant mice compared with controls, shown in Fig. 4A. While there was an increasing trend in mutant serum creatinine levels (0.36 ± 0.2 versus 0.30 ± 0.01 mg/dL, p = 0.17) and creatinine clearance (0.099 ± 0.01 versus 0.138 ± 0.02 mL/min, p = 0.09), we detected no significant changes in mutant mice versus controls (Fig. 4B and not shown). We detected no changes in the fractional excretion of sodium (0.0056 ± 0.002 versus 0.0048 ± 0.002%, p = 0.4) in the mutant mice versus controls. Comparing controls with mutants, we observed no statistically significant changes in serum sodium (153 ± 1 versus 154 ± 1 mEq/L), potassium (7.5 ± 0.2 versus 7.8 ± 0.3 mEq/L), chloride (113 ± 1 versus 114 ± 1 mEq/L), bicarbonate (28 ± 1 versus 26 ± 1 mM), or calcium (11 ± 0.2 versus 11 ± 0.1 mEq/L). Likewise, in comparing controls with mutants we observed no statistically significant changes in 24 h urine sodium (49 ± 7 versus 33 ± 5 mEq/L), calcium (5 ± 0.3 versus 4 ± 1.0 mEq/L), creatinine (22.8 ± 3.2 versus 14.7 ± 3.4 mg/dL), or Osmols (590 ± 85 versus 476 ± 88 mosmol/kg).

Renal function in mutant and control mice. Mutant (Mut) mice (n = 6) had significant increases in serum BUN levels (A) but no changes in serum creatinine levels (B) compared with the control (Con) mice (n = 8). Data shown are mean and SEM. * statistically significant from control, p < 0.05.
In comparing the kidneys at the time of sacrifice, we noted that one mutant mouse developed unilateral hydronephrosis, while the all other mutant and controls appeared grossly normal (data not shown). Mean kidney weights between the controls (n = 16) (0.21 ± 0.02 g) and mutants (n = 12) (0.26 ± 0.1 g) were not significantly different. Even when removing the single hydronephrotic kidney weight from the calculation, the mutant mean kidney weight (n = 11) (0.18 ± 0.03 g) was still not significantly different from the control mean weight. We also performed H & E staining on paraffin kidney sections from both the mutants and controls. In comparison with the controls, the hydronephrotic mutant kidney demonstrated marked parenchymal atrophy (data not shown). In addition, the non-hydronephrotic mutant kidneys demonstrated proteinaceous renal casts in cortical and medullary tubules and regenerative tubules with cytoplasmic basophilia (Fig. 5B). Mutant glomeruli demonstrated nuclear crowding, thickening and hypercellularity of the glomerular tuft, dilatation of Bowman's space, and thickening of Bowman's capsule (Fig. 5D). Thus the mutant kidneys had histologic evidence of chronic renal disease that correlated with the increases in BUN relative to controls.

Renal histology in mutant and control mice. In comparison with the controls (A), mutant kidneys demonstrate proteinaceous renal tubular casts (arrowheads) and regenerative tubules with cytoplasmic basophilia (B). In comparison with controls (C), mutant glomeruli (D) demonstrate nuclear crowding, thickening and hypercellularity of the glomerular tuft, dilatation of Bowman's space, and thickening of Bowman's capsule (arrow). A,B = 200 × magnification; C,D = 400 × magnification. Scale bar for A,B = 50 μm. Scale bar for C,D = 50 μm.
DISCUSSION
LBW infants with reduced nephron numbers have a significantly increased risk for hypertension later in life, which is a significant health problem (4–6). While the reason(s) for the increased risk is not clear, reduction in nephron number and LBW have each been implicated (4–6). Optimal therapies for this condition are poorly defined and, as the incidence continues to rise, the importance and medical costs associated with this syndrome are likely to escalate.
In 1988, Brenner et al. proposed that reduced nephron number or “nephron underdosing” independently and inversely correlated to risk of developing hypertension in adulthood (7). While some recent clinical data supports this hypothesis, there is no clear consensus regarding the association between reduced nephron number and development of hypertension. Most of the experimental data employs unilateral nephrectomy models (e.g. sheep) to study the underlying mechanisms of hypertension (9–10). However, increasing evidence suggests that hypertension, although developed in adulthood are primed in fetal life (4) and therefore experimental models identifying the phenomenon in utero would greatly influence our understanding of the patho-physiologic mechanisms associated with the disease. Here we pursued the development of a relevant and convenient in utero mouse model with reduced nephron numbers. The data presented herein provide evidence that the mutant mouse model we used is an excellent model to study effects of in utero reduced nephron number alone on development of hypertension in adulthood and are consistent with observations in clinical settings.
Our data showing a link between a reduction of nephron number in utero and hypertension in adulthood is consistent with data in GDNF heterozygous mice (15). These mice experience a 30% reduction in nephron number during fetal life, which results in significant elevations in blood pressure at 14 mo of age. Although birth weights were not recorded, it was implied that there were no differences between mutants and controls. In our model, there is a 24% reduction in nephron number due to lower ureteric bud tips numbers, and despite no differences in birth weight, mutants have dramatic increases in blood pressure at one year of age. In our study, the increases in blood pressure were accompanied by significant cardiac remodeling. Overall heart mass/body mass, LV area, and LV wall-thickness were all increased in our mutant mice relative to controls, clearly indicating end-organ injury from hypertension. It is possible that the mutant mice in our study are hypertensive for some reason other than a reduction in nephron number. However, 2 independent mouse models (our mice and the GDNF heterozygotes) both develop a bilateral, symmetric reduction in nephron number and subsequent hypertension, strongly supporting that alteration in nephron number alone increases the risk of hypertension later in life.
Since hypertension must be associated with changes in cardiac output and/or systemic vascular resistance, we performed 2-dimensional echocardiography to clarify the changes in our model. We observed significant increases in systemic vascular resistance and a decreasing trend in cardiac output. In contrast, Mortiz et al. have associated increased blood pressure to increased cardiac output in sheep models of unilateral nephrectomy (10). One potential reason for the discrepancy between the studies may be related the timing of the data collection. A commonly accepted paradigm for primary hypertension is that early on, there is an increase in cardiac output and a normal total peripheral resistance, followed by adaptive changes causing and eventual reduction in cardiac output and increased total peripheral resistance (19). The differences may also be species dependent (sheep versus mice). Finally, while the sheep model is a unilateral nephrectomy model, the mice in this study never form the full complement of nephrons.
In addition to the changes in blood pressure, we observed significant increases in BUN levels in our mutant mice compared with controls. In contrast, there were no statistically significant differences in mutant creatinine levels versus controls. One potential explanation is that the mice had decreased effective intravascular volume (e.g. dehydration), which is often known to cause a disproportionate increase in BUN relative to creatinine (20). This is not likely given that adult mutant weights and fractional excretion of sodium measurements were not lower than controls (as would be expected with dehydration or intravascular volume depletion). The mutant mice more likely have mild intrinsic renal dysfunction as is strongly supported by the histologic evidence of glomerular and tubular injury in these mice. Although one would then expect increases in serum creatinine in mutants versus controls (as well as the increases in BUN), it may be that the intrinsic lab error in our creatinine measurements does not allow us to measure these differences.
In summary, we have shown that adult fgfr2 ureteric bud knock out mutant mice develop hypertension and we have established its role as a relevant in utero mouse model to study hypertension and chronic renal failure that develops in adult life. Further studies defining the link between adult hypertension, chronic renal failure, and prenatal loss of nephrons will provide important mechanistic insights and may reveal new therapeutic opportunities for these important complications.
Abbreviations
- BUN:
-
blood urea nitrogen
- fgfr2:
-
fibroblast growth factor receptor 2
- GDNF:
-
glial cell line-derived neurotrophic factor
- LV:
-
left ventricular
References
Heart and Stroke Facts. American Heart Association. Available at http://www.americanheart.org
Martin JA, Kochanek KD, Strobino DM, Guyer B, Macdorman MF 2005 Annual summary of vital statistics-2003. Pediatrics 115: 619–634
Manalich R, Reyes L, Herrera M, Melendi C, Fundora I 2000 Relationship between weight at birth and the number and size of renal glomeruli in humans: a histomorphometric study. Kidney Int 58: 770–773
Amann K, Plank C, Dotsch J 2004 Low nephron number- a new cardiovascular risk factor in children?. Pediatr Neprol 19: 1319–1323
Kistner A, Celsi G, Vanpee M, Jacobson SH 2005 Increased systolic daily ambulatory blood pressure in adult women born preterm. Pediatr Neprol 20: 232–233
Huxley RR, Shiell AW, Law CM 2000 The role of size at birth and postnatal catch–up growth in determining systolic blood pressure: a systematic review of the literature. J Hypertens 18: 815–831
Brenner BM, Garcia DL, Anderson S 1988 Glomeruli and blood pressure. Less of one, more of the other?. Am J Hypertens 1: 335–347
Keller G, Zimmer G, Mall G, Ritz E, Amann K 2003 Nephron number in patients with primary hypertension. N Engl J Med 348: 101–108
Moritz KM, Wintour EM, Dodic M 2002 Fetal uninephrectomy leads to postnatal hypertension and compromised renal function. Hypertension 39: 1071–1076
Moritz KM, Jefferies A, Wong J, Wintour EM, Dodic M 2005 Reduced renal reserve and increased cardiac output in adult female sheep uninephrectomized as fetuses. Kidney Int 67: 822–828
Bauer R, Walter B, Bauer K, Klupsch R, Patt S, Zwiener U 2002 Intrauterine growth restriction reduces nephron number and renal excretory function in newborn piglets. Acta Physiol Scand 176: 83–90
Zimanyi MA, Bertram JF, Black MJ 2004 Does a nephron deficit in rats predispose to salt-sensitive hypertension?. Kidney Blood Press Res 27: 239–247
Vehaskari VM, Aviles DH, Manning J 2001 Prenatal programming of adult hypertension in the rat. Kidney Int 59: 238–245
Bassan H, Trejo LL, Kariv N, Bassan M, Berger E, Fattal A, Gozes I, Harel S 2000 Experimental intrauterine growth retardation alters renal development. Pediatr Neprol 15: 192–195
Cullen-McEwen LA, Kett MM, Dowling J, Anderson WP, Bertram JF 2003 Nephron number, renal function, and arterial pressure in aged GDNF heterozygous mice. Hypertension 41: 335–340
Zhao H, Kegg H, Grady S, Truong HT, Robinson ML, Baum M, Bates C 2004 Role of fibroblast growth factor receptors 1 and 2 in the ureteric bud. Dev Biol 276: 403–415
Weinstein DM, Mihm MJ, Bauer JA 2000 Cardiac peroxynitrite formation and left ventricular dysfunction following doxorubin treatment in mice. J Pharmacol Exp Ther 294: 396–401
Chaves AA, Weinstein DM, Bauer JA 2001 Non-invasive echocardiographic studies in mice: influence of anesthetic regimen. Life Sci 69: 213–222
Wikstrand J 1984 Left ventricular function in early primary hypertension. Functional consequences of cardiovascular structural changes. Hypertension 61: 108–116
Sklar AH, Riesenberg LA, Ur Rehman A, Smith S, Rivera-Padilla H 1996 Prerenal azotemia: differentiation of hyperureagenesis from renal hypoperfusion using urinary urea nitrogen data. Int J Artif Organs 19: 164–169
Acknowledgements
The authors would like to thank Dr. Michael Robinson, Dr. Kirk McHugh, and Dr. Donna Kusewitt for helpful discussions.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Poladia, D., Kish, K., Kutay, B. et al. Link Between Reduced Nephron Number and Hypertension: Studies in a Mutant Mouse Model. Pediatr Res 59, 489–493 (2006). https://doi.org/10.1203/01.pdr.0000202764.02295.45
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/01.pdr.0000202764.02295.45
This article is cited by
-
Fibroblast growth factor receptor signaling in kidney and lower urinary tract development
Pediatric Nephrology (2016)
