
Abstract
The hyperinsulinism/hyperammonemia (HI/HA) syndrome is caused by “gain of function” of glutamate dehydrogenase (GDH). Several missense mutations have been found; however, cell behaviors triggered by the excessive GDH activity have not been fully demonstrated. This study was aimed to clarify electrophysiological mechanisms underlying the dysregulated insulin secretion in pancreatic beta cells with GDH mutations. GDH kinetics and insulin secretion were measured in MIN6 cells overexpressing the G446D and L413V. Membrane potentials and channel activity were recorded under the perforated-patch configuration that preserved intracellular environments. In mutant MIN6 cells, sensitivity of GDH to guanosine triphosphate (GTP) was reduced and insulin secretion at low glucose concentrations was enhanced. The basal GDH activity was elevated in L413V bearing a mutation in the antenna-like structure. The L413V cells were depolarized without glucose, often accompanying by repetitive Ca2+ firings. The depolarization was maintained in the presence of adenosine triphosphate (ATP) and disappeared by depleting ATP, suggesting that the depolarization depended on intracellular ATP. In L413V cells, the ATP-sensitive potassium channel (KATP channel) was suppressed and the nonselective cation channel (NSCC) was potentiated, while sensitivity of the channels to their specific blockers or agonists was not impaired. These data suggest that the L413V cells increase the intracellular ATP/adenosine diphosphate (ADP) ratio, which in turn causes sustained depolarization not only by closure of the KATP channel, but also by opening of the NSCC. The resultant activation of the voltage-gated Ca2+ channel appears to induce hyperinsulinism. The present study provides evidence that multiple channels cooperate in unregulated insulin secretion in pancreatic beta cells of the HI/HA syndrome.
Similar content being viewed by others


Main
The HI/HA syndrome is an autosomal dominant disorder characterized by symptomatic hypoglycemia and persistent high plasma ammonia levels (1,2). Inappropriate secretion of insulin in pancreatic beta cells and defective nitrogen metabolism in the liver are caused by defects of the gene encoding GDH. The pathogenesis of concurrent hyperinsulinism and hyperammonemia is dysregulation, in other words, “gain of function,” of GDH. Missense mutations were first discovered in 15 amino acid sequences between F440 and H454 (exons 11 and 12), within the pivot helix of GDH (site 1) (3,4). Recently, another two sites exhibiting gain of function have been identified: the site 2 mutation (exons 6 and 7) in the GTP-binding region (5,6) and the site 3 mutation (exon 10) in the α-helix of the antenna-like structure on the other side from the hinge (7,8). Although mitochondrial GDH is found in all organisms, this antenna region does not exist in bacterial or fungal GDH. So far, only two mutations in site 3 mutations have been reported: L413V (7) and N410T (8). We previously reported two GDH mutations in Japanese patients [L413V (site 3) and G446D (site 1)] and characterized allosteric regulation of the GDH activity in COS cells overexpressing the mutant genes (7). However, we have not examined the effect of the two mutations on the GDH kinetics and insulin secretion in pancreatic beta cells. Moreover, cell responses mediating the unregulated insulin secretion with the GDH mutations remain to be defined.
GDH oxidizes L-glutamate to α-ketoglutarate using nicotinamide adenine dinucleotide (NAD) and/or nicotinamide adenine dinucleotide phosphate (NADP) as a cofactor. Activated GDH enhances glutamate oxidation and hence generates α-ketoglutarate, a substrate for the tricarboxylic acid cycle. The resultant increase in the intracellular ATP/ADP ratio may increase exocytosis of insulin by closing the KATP channel. The KATP channel may play a crucial role in the impaired insulin secretion of the HI/HA syndrome because diazoxide, an activator for the channel, is the most effective drug to prevent hypoglycemia (2,3). The aim of the present study was to clarify the cellular events responsible for the unregulated insulin secretion introduced by GDH defects. We overexpressed two GDH mutations (G446D and L413V) in MIN6 cells and investigated the GDH kinetics, insulin secretion, and electrophysiological properties. Together with enhanced insulin secretion, transformed MIN6 cells were depolarized through overproduction of ATP. This depolarization led to repetitive openings of the voltage-gated L-type Ca2+ channel (VDCC). In addition to sustained closure of the KATP channel, we found that the NSCC was activated with the GDH mutation. These findings clearly show that excessive GDH activity triggers changes in the activity of multiple channels and serves as a key regulator for hyperinsulinism of the HI/HA syndrome.
MATERIALS AND METHODS
Preparation of the recombinant adenoviral vector.
Full-length human GDH (hGDH) cDNA from human hepatocytes was generously provided by Dr. N. Amuro of the Nippon Medical School (9). Mutant hGDH cDNA of L413V and G446D was synthesized by specific base substitutions using site-directed mutagenesis, as described previously (7). Recombinant adenoviral vectors (rAdV) were generated to express the cDNA using the FLP-FRT recombination system as described previously (10). The concentration of viral solution achieved finally at 4.5 × 109 (L413V), 3.1 × 1010 (G446D), and 1.6 × 1010 (Wild) plaque-forming units/mL.
Measurement of GDH activity.
MIN6 cells (11) were infected with rAdV at a multiplicity of infection of 30, which induced expression of foreign genes in nearly 100% of the cells (12). Forty-eight hours after the infection, cells were harvested and GDH activity was measured as described previously (7). We examined the transfection efficiency among Wild, L413V, and G446D. For Western blot analysis, the extracts were electrophoresed in sodium dodecylsulfate (SDS)-polyacrylamide gel and subsequently transferred to GeneScreen hybridization transfer membrane (Du-Point MEN, Boston, MA). Immunoreactive GDH was identified by using primary antibody of a rabbit anti-calf GDH antibody (Amersham Pharmacia Biotech, Uppsala, Sweden), secondary antibody of a goat anti-rabbit IgG-peroxidase (Roche Diagnostics, Mannheim, Germany), and ECL system (Amersham Pharmacia Biotech). To ensure that each lane was loaded with an equivalent amount of protein, the blots were stripped with 0.2 N NaOH and reprobed with monoclonal anti-β-actin antibody (Sigma Chemical Co., St. Louis, MO) as described above.
Analysis of insulin secretion.
Cells (1.5 × 105 cell/well, 48-well plate) were infected with rAdV at a multiplicity of infection of 30. Forty-eight hours later, insulin secretion was assayed by the static incubation method (13). In brief, after a 30-min preincubation in N-2-hydroxyethylpiperazine N1-2-ethanesulfonic acid (HEPES)-balanced Krebs-Ringer buffer (KRB) (in mmol/L) (119 NaCl, 4.7 KCl, 1.2 MgSO4, 1.2 KH2PO4, 25 NaHCO3, 2.5 CaCl2, 10 HEPES, pH 7.4) containing 0.2% bovine serum albumen (BSA) and 2 mmol/L glucose, the preincubation buffer was replaced with fresh HEPES-balanced KRB containing 0.2% BSA and various concentrations (2–15 mmol/L) of glucose in the absence or in the presence of effectors. After an additional incubation at 37°C for 2 h, the buffer was collected, and released insulin was measured by Insulin, Rat, ELISA kit (Mercodia, Uppsala, Sweden).
Electrophysiological analysis.
MIN6 cells were plated on 13-mm micro cover glasses (Matsunami Glass Co., Kishiwada, Japan) and incubated at 37°C for 24 h. On the following day, cells were cotransfected with expression plasmids for 0.3 μg L413V (or Wild) cDNA and pEGFP-C1 plasmid vector (Clontech, Palo Alto, CA) using Effectene Transfection Reagent (Qiagen, Hilden, Germany). Twenty-four hours after transfection, recordings were made in transformed cells identified by GFP fluorescence at room temperature (20–24°C). To maintain an intact intracellular environment, currents and potentials were recorded under the perforated-patch configurations using amphotericin B unless described otherwise (14). Intracellular diffusible molecules, including ATP, hardly pass through amphotericin pores. This allowed the cell machinery to be preserved during the recordings. Voltage and current signals were recorded with an amplifier (Axonpatch 200A; Axon Instruments, Foster City, CA), digitized at 2 kHz with an analog-digital converter (Digidata 1200; Axon Instruments), and analyzed using pCLAMP software (Axon Instruments). The borosilicate glass pipettes had a resistance of 10–15 MΩ. The reference electrode was an Ag-AgCl wire connected to the bath solution through a Ringer-agar bridge. The zero current potential before the formation of the gigaseal (1–5 GΩ) was taken as 0 mV. The membrane potential was measured under the current-clamp mode. The membrane current was recorded under the voltage-clamp mode with either voltage ramps (−120 to 40 mV/s) or voltage steps (in 20-mV increments) applied at a holding potential of −60 mV. The extracellular solution contained (in mmol/L): 135 NaCl, 5 KCl, 5 CaCl2, 2 MgSO4, and 5 HEPES-NaOH (pH 7.4). In most experiments, 30 mmol/L NaCl was replaced by 30 mmol/L tetraethylammonium (TEA), to reduce delayed rectifying K+ (Kv) currents (14). This amount of TEA inhibited inward KATP currents only slightly (<10%). The pipette solution contained (in mmol/L): 107 KCl, 11 EGTA, 2 MgSO4, 1 CaCl2, 11 HEPES-KOH (pH 7.2). Amphotericin B (500 μg/mL) was added to the pipette solution. In some experiments, cells were dialyzed intracellularly with the pipette solution by rupturing patch membrane (the whole-cell configuration).
Data presentation and statistical analyses.
Data were expressed as mean ± SEM. Statistical significance (p < 0.05) was determined by an unpaired t test.
RESULTS
GDH activity.
The basal GDH activity in MIN6 cells transfected with rAdV-Wild (98 ± 9 nmol/mg of protein/min, n = 6) was 3-fold higher than those with an endogenous background (Mock: 33 ± 2, n = 6) (Fig. 1 A and B). Hence, we evaluated the results by comparing mutant GDH (L413V, G446D) with Wild. The basal GDH activity of L413V (236 ± 15, n = 6) was 2-fold higher than that of Wild and G446D (92 ± 11, n = 6) under the same amount of cross-reacting immunologic material for anti-GDH antibody. Thus, it was proved that the elevation in the basal GDH activity of L413V was not caused by a difference in transfection efficiency but increased enzyme specific activity (Fig. 1C). Figure 1A shows that L413V and G446D had severe insensitivity for GTP inhibition: the GDH activity of Mock (33 ± 2) and Wild (98 ± 9) was inhibited and reached 0.8 ± 0.6 and 2.4 ± 0.9 nmol/mg of protein/min, respectively, at 1 μmol/L GTP. However, the mutant constructs were weakly inhibited by 1 μmol/L GTP from 236 ± 15 to 170 ± 20 in L413V and from 92 ± 11 nmol/L to 70 ± 12 in G446D. As for the allosteric effect of ADP (Fig. 1B), all the constructs finally reached the similar steady state at 100 μmol/L ADP. The half-maximal stimulatory concentrations of ADP in L413V and G446D (17 ± 2 and 20 ± 2 μmol/L, n = 3, respectively) did not differ from those in Mock and Wild (22 ± 4 and 18 ± 4 μmol/L, n = 3, respectively). These results indicate that the L413V mutation (site 3) shared common features of allosteric regulation by nucleotides with the G446D mutation (site 1), but had a marked elevation of the basal GDH activity.

Allosteric regulation of GDH activity in MIN6 cells transfected with the mutants and Wild GDH. The GDH activity at different concentrations of GTP (A) and ADP (B). Data are mean ± SEM (n = 3). **p < 0.005, *p < 0.05 for the comparison with rAdV-Wild constructs. (C) Western blot analysis of GDH protein in Mock cells and Min6 cells transfected with rAdV-Wild, L413V, and G446D constructs. Each single band in Mock, Wild, L413V, and G446D was shown at the expected size of 54 kD under the equivalent amount of β-actin.
Insulin secretion.
Glucose stimulated insulin secretion in a dose-dependent-manner from MIN6 cells transfected with the rAdV-L413V, -G446D, and -Wild GDH because of its high transfection efficiency (Fig. 2). In mutant cells, however, insulin secretion was enhanced at low concentrations of glucose (0–5 mmol/L). There was no significant difference in the secretion at higher concentrations of glucose (10–15 mmol/L). Furthermore, diazoxide, an activator for the KATP channel, decreased insulin secretion to 50%, and glibenclamide, a blocker for the KATP channel, increased insulin secretion by 2.0- to 2.5-fold. These data indicate that functional KATP channels are well preserved in mutant cells and contribute to insulin secretion in the mutant cells. In addition, the higher basal GDH activity of L413V compared with that of G446D seemed not to affect the mode of up-regulation of insulin secretion.

Insulin secretion of MIN6 cells transfected with the mutants and Wild GDH under the various concentrations of glucose (0–15 mmol/L). Effects of the KATP channel effectors of diazoxide (200 μmol/L) and 500 nmol/L glibenclamide were examined at 5 and 2 mmol/L glucose, respectively. The amount of insulin secretion was normalized by the cellular protein content in each well. Insulin secretion in control Wild cells at 2 mmol/L glucose was standardized to 1.0. Each assay (n = 7–10) was performed in triplicate. *p < 0.01.
Electrophysiological properties.
In the preliminary experiments, we compared the two expression procedures of the rAdV and the lipofection-gene transfer methods. The membrane potentials in MIN6 cells with L413V were depolarized at almost the same levels using both methods in comparison with control MIN6 cells. Identification of well-transfected cells was attained easily by cotransfection of GDH gene and GFP fluorescence using the lipofection method. Therefore, this method was selected because electrophysiological studies were conducted on single transfected cells. Figure 3A shows representative currents recorded from Wild and L413V cells under the perforated-patch configuration. Glucose was omitted from the extracellular medium to maximize the effects of the GDH mutation on insulin secretion. Repetitive inward currents were evoked by depolarization in L413V cells, but not in Wild cells. The membrane potential of MIN6 cells (Mock) was −71.6 ± 0.8 mV (n = 3) under physiological conditions (Fig. 3 B). Addition of TEA (30 mmol/L), a blocker of the Kv channel, depolarized both Mock (data not shown) and Wild cells, but still did not elicit firings (Fig. 3B). L413V cells had more positive membrane potentials and were often accompanied by repetitive firings (Fig. 3B). The firing was likely due to VDCC because they were completely blocked by 100 nmol/L nifedipine, a blocker of VDCC. Thus, L413V sustained depolarization, which could activate VDCC. The membrane potential of L413V cells (−0.1 ± 1.7 mV, n = 22) was significantly higher than that of Mock cells (−30.0 ± 7.8 mV, n = 7) and Wild cells (−27.0 ± 3.4 mV, n = 15) (Fig. 3C) in the presence of TEA.

Membrane currents and potentials in MIN6 cells overexpressing L413V. (A) Currents evoked by voltage steps applied at a holding potential of −60 mV under the perforated-patch configuration in a Wild cell and a L413V cell. (B) Membrane potentials of a Mock cell recorded under the physiological solution and of Wild and L413V cells in the presence of TEA. (C) Membrane potentials of MIN6 cells (Mock, n = 7; Wild, n = 15; L413V, n = 22). The extracellular medium contained 30 mmol/L TEA. All recordings in this and subsequent figures were made in the absence of glucose. **p < 0.001.
The enhanced GDH activity may elevate the intracellular ATP through increased glutamate oxidation. To examine the involvement of ATP in the depolarization of transformed cells, the inside of L413V cells were dialyzed with ATP-omitting and -containing solutions by rupture of the patch membrane (whole cell configuration) (Fig. 4). The depolarization was maintained in the presence of ATP (−4.2 ± 5.2 mV, n = 3) but disappeared by depleting ATP. In the absence of ATP, there was no significant difference in the membrane potential between L413V (−63.0 ± 2.0 mV, n = 3) and Wild (−61.6 ± 0.8 mV, n = 3) cells. These results suggest that intracellular ATP is a critical regulator for the membrane potential and that excess ATP may generate the sustained depolarization in L413V cells.

Effects of intracellular ATP on membrane potentials of MIN6 cells. L413V cells under perforated-patch configuration are shown on the left. The membrane potentials recorded in the whole-cell configuration following rupture of the patch membrane in L413V cells dialyzed intracellularly with either ATP (5 mmol/L)-containing or ATP-free pipette solutions and in Wild cells, with an ATP-free solution are shown on the right. The extracellular medium was Ringer solution. (n = 3–6). **p < 0.001.
Figure 5A shows the current-voltage relationships obtained by voltage ramps. The currents contained KATP currents because they were reduced by glibenclamide and increased by diazoxide. The reversal potential of the KATP current was −50.9 ± 2.7 mV (n = 10). We evaluated the KATP channel activity as the glibenclamide-sensitive component. The current density was measured at −100 mV, at which the Kv channel is closed. (Fig 5C) The KATP current in L413V cells (−1.02 ± 0.35 pA/pF, n = 9) was significantly lower than that in Wild cells (−2.30 ± 0.37 pA/pF, n = 8).

KATP channels and NSCC. (A) Current-voltage (I-V) relationships of MIN6-Wild cells evoked by voltage ramps applied at −60 mV before and after addition of 500 nmol/L glibenclamide (Glib), and 200 μmol/L diazoxide (Diaz). (B) Current-voltage I-V relationships before and after addition of 10 nmol/L GdCl3 and 100 pmol/L maitotoxin. (C) Glibenclamide-sensitive currents (KATP) and GdCl3-sensitive currents (NSCC) in MIN6-Wild and L413V cells. The ordinate represents current density at −100 mV. Cell capacitance: 6.3 ± 0.9 pF (n = 8) in Wild cells and 6.8 ± 0.4 pF (n = 9) in L413V cells. *p < 0.01.
In addition to KATP channels, NSCC may play a role in insulin secretion in pancreatic beta cells by regulating membrane potential and thereby glucose signaling (15). The currents of MIN6 cells were decreased by an NSCC blocker, GdCl3 (Fig. 5B) and activated by an NSCC activator, maitotoxin (16). The GdCl3- and maitotoxin-sensitive current had a reversal potential of −6.6 ± 1.5 mV (n = 10) and thus was most likely an NSCC current. The NSCC current, estimated as the GdCl3-sensitive component, was significantly greater in L413V cells (−3.46 ± 0.42 pA/pF, n = 9) than those of Wild cells (−2.06 ± 0.16 pA/pF, n = 8) (Fig. 5C). Thus, it is deemed that both closure of the KATP channel and activation of NSCC contribute to the sustained depolarization in L413V cells.
DISCUSSION
Enhanced GDH activity and increased insulin secretion are essential features of the HI/HA syndrome, although mutations occur at various sites of the gene encoding GDH, such as site 1 for the pivot helix (3,4), site 2 for interaction with GTP molecules (5,6), and site 3 for the antenna-like structure (7,8). With all these GDH mutations, allosteric inhibition of GDH by GTP is severely impaired, but allosteric excitation by ADP is intact. A dose-dependent up-regulation of glucose-induced insulin secretion was reported in a Y266C mutation (site 2) (13,17); potentiation of secretion is evident only at low glucose level. On the other hand, Wild GDH overexpression in rat islets does not change insulin release at basal or intermediate glucose but stimulates insulin secretion by high glucose concentrations, glutamine, and 2-aminobicyclo-[2,2,1] heptane-2-carboxylic acid compared with controls (18). The present study revealed that both G446D and L413V mutations also caused higher insulin release at low glucose concentrations (0–5 mmol/L). There was no difference in the maximum amount of insulin secreted at higher glucose levels in the GDH mutations. Thus, various GDH mutations share common phenotypes of impaired GDH kinetics and unregulated insulin secretion. However, the L413V mutation was distinct in its high basal GDH activity in MIN6 cells. We reported similarly high GDH activity in lymphoblasts of L413V patients and COS cells transformed with L413V constructs (7). Elevation in the basal GDH activity was also reported in a N410T GDH mutation (site 3) (8). The difference in the in vitro basal GDH activity between L413V and G446D had little effect on the mode of insulin secretion. We may suspect that the different basal GDH activities do not affect insulin section and that the summation and balance of allosteric regulation on in vivo GDH are more important for insulin secretion than the basal GDH activity.
Although the list of GDH mutations is increasing, we do not have a clear picture of cell behavior during the process leading to unregulated insulin secretion in beta cells bearing the GDH gene defects. Intracellular dialysis with ATP-containing and -omitting solutions revealed that the depolarization in L413V cells was mimicked by introduction of 5 mmol/L ATP, which could block the KATP channel sufficiently (19). Therefore, the amount of ATP produced by the enhanced GDH activity may be enough to block the KATP channel and to secrete insulin even in the absence of glucose. Suppression of the KATP channel activity with GDH mutations was confirmed here using electrophysiology; the channel activity was decreased to less than half of that in Wild cells.
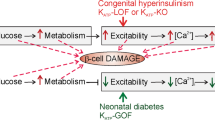
The KATP channel, composed of a sulfonylurea receptor (SUR1) (20) and an inward rectifier K+ channel (Kir 6.2) (19), is a key factor for insulin secretion in pancreatic beta cells. Mutations of KATP channels induce a variety of disturbances in glucose metabolism, such as persistent hyperinsulinemic hypoglycemia of infancy (PHHI) (21) and neonatal diabetes (22). PHHI is an autosomal disorder with mutations in SUR1 and Kir6.2. Although both the resting potential and the basal intracellular free Ca2+ concentration ([Ca2+]i) are elevated in Kir6.2 null mice (23) and SUR1 null mice (24), these animals remain euglycemic for a large portion of their life despite constant depolarization of membrane. Insulin release at low glucose of <2.8 mmol/L is not enhanced, but glucose-induced insulin secretion is lacking and agents acting on SUR1 are ineffective for regulation of insulin secretion. The HI/HA syndrome differs from PHHI in that functions of the KATP channel are intact: i.e. glucose-induced insulin secretion is dose dependent, although the dose-response curve shifts to a lower glucose level. The present study revealed that depolarization of the GDH-mutant beta cells is enough to evoke repetitive firings of VDCC, even in the absence of glucose. Functional KATP channels still play a key role in insulin secretion in mutant cells; therefore, diazoxide and glibenclamide efficiently regulate insulin secretion. Gain of function of GDH activity thus has the unique pathology responsible for unregulated insulin secretion.
Potentiation of NSCC is a novel feature accompanying GDH mutations, although this is unlikely to be specific to expression of mutant GDH. NSCC was first described in insulin-secreting cells (the CRI-GI cell line) (25) and recently attracted attention as a channel to regulate insulin secretion (15,16,26). Mutations of GDH did not alter the intrinsic property of the channel, such as the reversal potential and the sensitivity to GdCl3 and maitotoxin. Inositol 1,4,5-trisphosphate-induced Ca2+ mobilization from the intracellular Ca2+ stores (27), which mediates maitotoxin-induced activation of NSCC (28), may be intact as well. Activation of NSCC might contribute to either depolarization and/or elevation of [Ca2+]i because cations including Ca2+ permeate the channel (29). Both ATP and ADP are reported to inhibit NSCC, but an increase in the ATP/ADP ratio might reduce the inhibition of NSCC because ADP is a more potent inhibitor than ATP (16,25). Activation of maitotoxin-sensitive NSCC currents is inhibited by prolonged dialysis of cells with ATP-free solutions (16). Otherwise, the rise of [Ca2+]i due to a Ca2+ influx through activated VDCC may open NSCC, as NSCC are activated by increases in [Ca2+]i (30).
From the present findings, we can depict the sequential responses triggered by the enhanced GDH activity in the mutant beta cells of the HI/HA syndrome, i.e. excessive GDH activity elevates the intracellular ATP/ADP ratio, which in turn closes KATP channels and possibly opens NSCC. These changes in the channel activities lead to depolarization and subsequent activation of VDCC. The resultant elevation of [Ca2+]i reinforces the exocytotic secretion of insulin. The channels and cell mechanisms required for insulin secretion seem to maintain their intrinsic properties. MIN6 cells transfected with the GDH mutations provide a useful model system for understanding the pathogenesis of the HI/HA syndrome and for exploring treatments to cure insulin secretion disorders.
Abbreviations
- [Ca2+]i:
-
intracellular free Ca2+ concentration
- GDH:
-
glutamate dehydrogenase (EC 1.4.1.3)
- HI/HA:
-
hyperinsulinism/hyperammonemia
- KATP channel:
-
adenosine triphosphate–sensitive potassium channel
- Kir 6.2:
-
inward rectifier K+ channel
- NSCC:
-
nonselective cation channel
- PHHI:
-
persistent hyperinsulinemic hypoglycemia of infancy
- rAdV:
-
recombinant adenoviral vector
- SUR1:
-
sulfonylurea receptor
- TEA:
-
tetraethylammonium
- VDCC:
-
voltage-gated L-type Ca2+ channel
References
Zammarchi E, Filippi L, Novembre E, Donati MA 1996 Biochemical evaluation of a patient with a familial form of leucine-sensitive hypoglycemia and concomitant hyperammonemia. Metabolism 45: 957–960
Weinzimer SA, Stanley CA, Berry GT, Yudkoff M, Tuchman M, Thornton PS 1997 A syndrome of congenital hyperinsulinism and hyperammonemia. J Pediatr 130: 661–664
Stanley CA, Lieu YK, Hsu BY, Burlina AB, Greenberg CR, Hopwood NJ, Perlman K, Rich BH, Zammarchi E, Poncz M 1998 Hyperinsulinism and hyperammonemia in infants with regulatory mutations of the glutamate dehydrogenase gene. N Engl J Med 338: 1352–1357
Stanley CA, Fang J, Kutyna K, Hsu BY, Ming JE, Glaser B, Poncz M 2000 Molecular basis and characterization of the hyperinsulinism/hyperammonemia syndrome: predominance of mutations in exons 11 and 12 of the glutamate dehydrogenase gene. Diabetes 49: 667–673
De Lonlay P, Benelli C, Fouque F, Ganguly A, Aral B, Dionisi-Vici C, Touati G, Heinrichs C, Rabier D, Kamoun P, Robert JJ, Stanley C, Saudubray JM 2001 Hyperinsulinism and hyperammonemia syndrome: report of twelve unrelated patients. Pediatr Res 50: 353–357
Miki Y, Taki T, Ohura T, Kato H, Yanagisawa M, Hayashi Y 2000 Novel missense mutations in the glutamate dehydrogenase gene in the congenital hyperinsulinism hyperammonemia syndrome. J Pediatr 136: 69–72
Fujioka H, Okano Y, Inada H, Asada M, Kawamura T, Hase Y, Yamano T 2001 Molecular characterisation of glutamate dehydrogenase gene defects in Japanese patients with congenital hyperinsulinism/hyperammonaemia. Eur J Hum Genet 9: 931–937
Yorifuji T, Muroi J, Uematsu A, Hiramatsu H, Momoi T 1999 Hyperinsulinism-hyperammonemia syndrome caused by mutant glutamate dehydrogenase accompanied by novel enzyme kinetics. Hum Genet 104: 476–479
Amuro N, Goto Y, Okazaki T 1990 Isolation and characterization of the two distinct genes for human glutamate dehydrogenase. Biochim Biophys Acta 1049: 216–218
Tashiro F, Niwa H, Miyazaki J 1999 Constructing adenoviral vectors by using the circular form of the adenoviral genome cloned in a cosmid and the Cre-loxP recombination system. Hum Gene Ther 10: 1845–1852
Miyazaki J, Araki K, Yamato E, Ikegami H, Asano T, Shibasaki Y, Oka Y, Yamamura K 1990 Establishment of a pancreatic beta cell line that retains glucose-inducible insulin secretion: special reference to expression of glucose transporter isoforms. Endocrinology 127: 126–132
da Silva Xavier G, Varadi A, Ainscow EK, Rutter GA 2000 Regulation of gene expression by glucose in pancreatic beta-cells (MIN6) via insulin secretion and activation of phosphatidylinositol 3′-kinase. J Biol Chem 275: 36269–36277
Tanizawa Y, Nakai K, Sasaki T, Anno T, Ohta Y, Inoue H, Matsuo K, Koga M, Furukawa S, Oka Y 2002 Unregulated elevation of glutamate dehydrogenase activity induces glutamine-stimulated insulin secretion: identification and characterization of a GLUD1 gene mutation and insulin secretion studies with MIN6 cells overexpressing the mutant glutamate dehydrogenase. Diabetes 51: 712–717
Smith PA, Sellers LA, Humphrey PP 2001 Somatostatin activates two types of inwardly rectifying K+ channels in MIN-6 cells. J Physiol 532: 127–142
Qian F, Huang P, Ma L, Kuznetsov A, Tamarina N, Philipson LH 2002 TRP genes: candidates for nonselective cation channels and store-operated channels in insulin-secreting cells. Diabetes 51: S183–S189
Leech CA, Habener JF 1998 A role for Ca2+-sensitive nonselective cation channels in regulating the membrane potential of pancreatic beta-cells. Diabetes 47: 1066–1073
Anno T, Uehara S, Katagiri H, Ohta Y, Ueda K, Mizuguchi H, Moriyama Y, Oka Y, Tanizawa Y 2004 Overexpression of constitutively activated glutamate dehydrogenase induces insulin secretion through enhanced glutamate oxidation. Am J Physiol Endocrinol Metab 286: E280–E285
Carobbio S, Ishihara H, Fernandez-Pascual S, Martin-Del-Rio R, Maeshler P 2004 Insulin secretion profiles are modified by overexpression of glutamate dehydrogenase in pancreatic islets. Diabetologia 47: 266–276
Sakura H, Ammälä C, Smith PA, Gribble FM, Ashcroft FM 1995 Cloning and functional expression of the cDNA encoding a novel ATP-sensitive potassium channel subunit expressed in pancreatic beta-cells, brain, heart and skeletal muscle. FEBS Lett 377: 338–344
Aguilar-Bryan L, Nichols CG, Wechsler SW, Clement JP 4th, Boyd AE 3rd, González G, Herrera-Sosa H, Nguy K, Bryan J, Nelson DA 1995 Cloning of the beta cell high-affinity sulfonylurea receptor: a regulator of insulin secretion. Science 268: 423–426
Thomas P, Ye Y, Lightner E 1996 Mutation of the pancreatic islet inward rectifier Kir6.2 also leads to familial persistent hyperinsulinemic hypoglycemia of infancy. Hum Mol Genet 5: 1809–1812
Gloyn AL, Pearson ER, Antcliff JF, Proks P, Bruining GJ, Slingerland AS, Howard N, Srinivasan S, Silva JM, Molnes J, Edghill EL, Frayling TM, Temple IK, Mackay D, Shield JP, Sumnik Z, van Rhijn A, Wales JK, Clark P, Gorman S, Aisenberg J, Ellard S, Njolstad PR, Ashcroft FM, Hattersley AT 2004 Activating mutations in the gene encoding the ATP-sensitive potassium-channel subunit Kir6.2 and permanent neonatal diabetes. N Engl J Med 350: 1838–1849
Miki T, Nagashima K, Tashiro F, Kotake K, Yoshitomi H, Tamamoto A, Gonoi T, Iwanaga T, Miyazaki J, Seino S 1998 Defective insulin secretion and enhanced insulin action in KATP channel-deficient mice. Proc Natl Acad Sci U S A 95: 10402–10406
Shiota C, Larsson O, Shelton KD, Shiota M, Efanov AM, Hoy M, Lindner J, Kooptiwut S, Juntti-Berggren L, Gromada J, Berggren PO, Magnuson MA 2002 Sulfonylurea receptor type 1 knock-out mice have intact feeding-stimulated insulin secretion despite marked impairment in their response to glucose. J Biol Chem 277: 37176–37183
Sturgess NC, Hales CN, Ashford ML 1986 Inhibition of a calcium-activated, non-selective cation channel, in a rat insulinoma cell line, by adenine derivatives. FEBS Lett 208: 397–400
Suzuki M, Murata M, Ikeda M, Miyoshi T, Imai M 1998 Primary structure and functional expression of a novel non-selective cation channel. Biochem Biophys Res Commun 242: 191–196
Taylor CW, Laude AJ 2002 IP3 receptors and their regulation by calmodulin and cytosolic Ca2+. Cell Calcium 32: 321–334
Leech CA, Habener JF 1997 Insulinotropic glucagon-like peptide-1-mediated activation of non-selective cation currents in insulinoma cells is mimicked by maitotoxin. J Biol Chem 272: 17987–17993
Albert AP, Large WA 2002 A Ca2+-permeable non-selective cation channel activated by depletion of internal Ca2+ stores in single rabbit portal vein myocytes. J Physiol 538: 717–728
Reale V, Hales CN, Ashford ML 1994 Nucleotide Regulation of a calciumactivated cation channel in the rat insulinoma cell line, CRI-G1. J Membr Biol 141: 101–112
Author information
Authors and Affiliations
Corresponding author
Additional information
This study was supported in part by grants from the Ministry of Education, Culture, Sports, Science, and Technology of Japan; by grants from the Ministry of Health Sciences and Welfare of Japan; and by funds from Suyama Research Foundation.
Rights and permissions
About this article
Cite this article
Kawajiri, M., Okano, Y., Kuno, M. et al. Unregulated Insulin Secretion by Pancreatic Beta Cells in Hyperinsulinism/Hyperammonemia Syndrome: Role of Glutamate Dehydrogenase, ATP-Sensitive Potassium Channel, and Nonselective Cation Channel. Pediatr Res 59, 359–364 (2006). https://doi.org/10.1203/01.pdr.0000198775.22719.46
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/01.pdr.0000198775.22719.46
This article is cited by
-
Non-steroidal anti-inflammatory drugs increase insulin release from beta cells by inhibiting ATP-sensitive potassium channels
British Journal of Pharmacology (2007)
