
Abstract
In this study, we report on three Japanese patients with cytochrome P450 oxidoreductase (POR) deficiency (PORD). Case one was a 46,XY patient who was found to have mildly increased 17α-hydroxyprogesterone (17-OHP) by the neonatal mass screening. There was no maternal virilization during pregnancy, and he had no skeletal or genital abnormality. Thus, he was initially diagnosed as having nonclassical 21-hydroxylase deficiency (21-OHD). Cases two and three were 46,XX patients who were identified because of severely virilized external genitalia and maternal virilization during pregnancy. In case two, the neonatal mass screening was normal, and she had no skeletal abnormality except for mild adduction of bilateral third toes. Thus, she was initially diagnosed as having aromatase deficiency. In case three, the neonatal mass screening showed moderately increased 17-OHP, and no skeletal lesion other than rigid second metacarpophalangeal joints was identified in early infancy. Thus, she was initially suspected as having 21-OHD and/or aromatase deficiency. Subsequently, endocrine studies including urine steroid hormone analysis were performed for the assessment of glucocorticoid treatment in case one and for the virilized genitalia in cases two and three, showing adrenal and/or gonadal dysfunction characteristic of PORD. Thus, molecular analysis of POR was carried out, demonstrating homozygosity for R457H in cases one through three. The results imply that clinical features in PORD can be similar to those in 21-OHD or aromatase deficiency, and that comprehensive assessment of the pregnant course, physical examination, and adrenal and gonadal function studies is essential for the precise diagnosis of PORD.
Similar content being viewed by others

Main
Cytochrome P450 oxidoreductase (POR) deficiency (PORD) is a recently established autosomal recessive disorder caused by mutations in the gene encoding an electron donor of all microsomal P450 enzymes (1–3). Clinical features of this condition include skeletal malformations referred to as Antley-Bixler syndrome (ABS) and insufficient glucocorticoid production with increased 17α-hydroxyprogesterone (17-OHP) in patients of both sexes, normal to poor masculinization during fetal and pubertal periods in male patients, and virilization during fetal life and poor pubertal development without worsening of virilization in female patients, together with maternal virilization and low estriol (E3) during pregnancy (3–5). In addition, polycystic ovary (PCO) is frequently observed in affected females (1,2,4). Such clinical features are primarily explained by impaired activities of CYP51A1 (lanosterol 14α-demethylase) involved in cholesterologenesis and of CYP21A2 (21-hydroxylase), CYP17A1 (17α-hydroxylase and 17,20 lyase), and CYP19A1 (aromatase) involved in steroidogenesis (3,4,6).
Clinical features in PORD are highly variable among patients. For example, skeletal manifestation ranges widely from typical ABS to nearly normal findings (1,3,4). Thus, some patients may have clinical phenotype similar to that of 21-hydroxylase deficiency (21-OHD) characterized by steroidogenic impairment with increased 17-OHP in patients of both sexes and virilization in female patients (7). Similarly, other patients may exhibit clinical phenotype similar to that of aromatase deficiency associated with virilization and gonadal dysfunction with PCO in female patients and maternal virilization and low E3 during pregnancy (8,9). Here, we report on previously unreported three patients with PORD who were initially regarded as having nonclassical 21-OHD and/or aromatase deficiency.
METHODS
Patients.
Case one was the 2.82 kg (–0.4 SD) product born to unrelated and healthy parents at 38 wk of gestation in a local hospital. There was no maternal virilization during the pregnancy. Immediately after birth, he received surgical treatment for imperforate anus. There was no skin pigmentation or external genital abnormality. Since the neonatal mass screening performed at 23 d of age after the operation showed a mildly elevated 17-OHP (20.1 nmol/L) (cut off value, 10.0 nmol/L), he was referred to Jikei University Hospital. Blood endocrine studies suggested the diagnosis of nonclassical 21-OHD, and glucocorticoid replacement therapy was started from four months of age. The karyotype was 46,XY. Subsequently, urine steroid profile analysis was performed for the assessment of glucocorticoid therapy, revealing combined CYP21A2 and CYP17A1 deficiency. After the molecular diagnosis of PORD at 4.5 y of age, bone survey was performed, showing no discernible skeletal abnormalities. On the last examination at 4 y and 10 mo of age, he showed normal growth and development, and did not experience adrenal failure on the glucocorticoid supplementation therapy with the hydrocortisone dosage being gradually tapered from 10 to 3 mg/m2/d during the 4.5 y.
Case two was born to non-consanguineous and healthy parents at 39 wk of gestation with a birth length of 47.7 cm (–0.5 SD) and a birth weight of 2.54 kg (–1.3 SD) at a local hospital. Maternal virilization was noticed at the third trimester, and urine E3 measured only once at 35 wk of gestation was found to be low (30 μM) (normal range, 35–180 μM). External genitalia were severely virilized (Prader stage 5) (10) with a phallus-like structure of 1.0 cm long and complete labial fusion (Fig. 1). The meatus opened at the tip of the phallus-like structure. There was no skin pigmentation. The neonatal mass screening at five days of age was normal, and chromosome analysis revealed a 46,XX karyotype. Thus, she was initially diagnosed as having aromatase deficiency and was reared as a female. She was referred to National Center for Child Health and Development, and endocrine studies showed combined CYP21A2 and CYP17A1 and ovarian dysfunction. Thus, PORD was suspected and confirmed by molecular analysis. Subsequent physical examination revealed no demonstrable somatic abnormality indicative of skeletal lesion except for mild adduction of bilateral third toes. At three months of age, magnetic resonance imaging delineated bilateral PCO. She received genitoplasty at 18 mo of age. On the last examination at five years old, she showed normal growth and development, and had no adrenal crisis under no medication.

Severely virilized external genitalia (Prader stage 5) in two 46,XX patients with PORD.
Case three was born to non-consanguineous and healthy parents at 37 wk of gestation at a local hospital. The mother showed virilization at the third trimester. The birth length was 49.6 cm (+1.2 SD), the birth weigh 3.06 kg (+1.0 SD), and the birth head circumference 33.6 cm (+0.6 SD). Physical examination showed severely virilized external genitalia (Prader stage 5) with a phallus-like structure of 2.0 cm long and complete labial fusion (Fig. 1). The meatus opened at the tip of the phallus-like structure. There was no skin pigmentation. The neonatal mass screening at five days of age showed moderately elevated 17-OHP (56.5 nmol/L), and chromosome analysis showed a 46,XX karyotype. Thus, she was initially suspected as having 21-OHD and/or aromatase deficiency, and was raised as a female. Although some rigidity was noticed at the bilateral second metacarpophalangeal joints, it was regarded as a co-incidental phenotype. She was referred to Keio University Hospital, and PORD was suspected and confirmed by urine steroid profile analysis and molecular analysis. At two months of age, bilateral PCO was identified by magnetic resonance imaging. At four months of age, although her head circumference was normal (41.5 cm, +0.3 SD), premature closure of anterior fontanel was noted, and a bone survey indicated the tendency of craniosynostosis and bilateral arachnodactyly. At 12 mo of age, she showed normal growth and development, and had no episode of adrenal crisis under no medication. She has been planned to receive genitoplasty at 1.5 y of age.
Endocrine studies.
Serum adrenal steroids were measured before and after ACTH stimulation (250 μg/m2 bolus IV; blood sampling at 0 and 60 min) in cases one through three. In cases two and three, basal and peak serum LH and FSH were obtained during a gonadotropin releasing hormone stimulation test (100 μg/m2 bolus IV; blood sampling at 0, 30, 60, 90, and 120 min), and serum testosterone (T) was determined before and after a human chorionic gonadotropin stimulation (3,000 IU/m2/dose IM for three consecutive days; blood sampling on days one and four), to examine the possibility of true hermaphroditism. In addition, basal plasma ACTH and renin activity were obtained in cases one through three, and basal serum estradiol was measured in cases two and three. The results were compared with the age-matched reference data (11–13). Furthermore, cortisol and 17-OHP values were also compared with those of patients with classical and/or nonclassical 21-OHD (14).
Urine steroid hormone metabolite (M) profile was determined for random urine samples by a gas chromatograph mass spectrometry (15). The results were compared with our data obtained from the following 1,111 subjects born at term: 1) 914 healthy subjects (3–914 d of age); 2) 95 subjects with transient elevation of serum 17-OHP (3–699 d of age); 3) 92 subjects with classical 21-OHD before treatment (3–116 d of age); and 4) 10 subjects with non-classical 21-OHD before treatment (13–368 d of age). Transient elevation of serum 17-OHP was defined as a condition in which serum 17-OHP was normalized by 90 d of age. Classical and non-classical 21-OHD was defined by the clinical criteria (16).
Mutation analysis of POR.
This study has been approved by the Institutional Review Board Committee at National Center for Child Health and Development. After taking written informed consent, leukocyte genomic DNA was obtained from cases one through three and the parents of cases two and three. Direct sequencing was performed for the 15 exons and their flanking introns of POR, as previously described (4).
RESULTS
Endocrine studies.
Blood hormone data were compatible with defective adrenal and gonadal steroidogenesis (Table 1). ACTH was mildly elevated, and cortisol was normal at baseline but did not increase to the normal range after ACTH stimulation. This blunt cortisol response was similar to that of nonclassical 21-OHD. Basal and ACTH stimulated 17-OHP values were grossly similar to those of nonclassical 21-OHD and lower than those of classical 21-OHD. Pregnenolone and progesterone were elevated, and 17-OH pregnenolone was normal or elevated. Basal and ACTH stimulated deoxycorticosterone (DOC), dehydroepiandrosterone (DHEA), and androstenedione (Δ4A) were grossly normal, as were basal renin activity and aldosterone. Basal and gonadotropin releasing hormone stimulated LH and FSH were normal or elevated, whereas basal and human chorionic gonadotropin stimulated T as well as basal estradiol were normal or decreased.
Urine hormone data were primarily consistent with combined CYP21A2 and CYP17A1 deficiency (Fig. 2). Progesterone-M, 17-OHP-M, and 21-deoxycortisol (DOF)-M values tended to be higher than those in subjects with transient elevation of serum 17-OHP and control subjects; furthermore, progesterone-M values were similar to those in subjects with classical and non-classical 21-OHD, 17-OHP-M values were similar to those in subjects with nonclassical 21-OHD and lower than those in subjects with classical 21-OHD, and 21-DOF-M values were lower than those in subjects with classical and non-classical 21-OHD. DHEA-M values tended to be lower than those in nonPORD subjects, whereas Δ4A-M values, especially those before four months of age, tended to be elevated with some overlap with those in subjects with transient elevation of serum 17-OHP and classical and non-classical 21-OHD; of Δ4A-Ms, Δ4A-M (5α-reduced) (androsterone) showed a similar trend, while Δ4A-M (5β-reduced) (etiocholanolone) remained grossly normal. 11-OHΔ4A-M (5α-reduced) (11-hydroxyandrosterone) values were normal or decreased, which were contrastive to high values in subjects with classical and nonclassical 21-OHD; thus, the ratios between 11-OHΔ4A-M (5α-reduced) and progesterone-M were low in cases one through three with PORD and high in subjects with classical and nonclassical 21-OHD. 17-OH pregnenolone-M, aldosterone-M, and cortisol-M values were similar among all subjects examined including cases one through three. Pregnenolone-M, DOC-M, and corticosterone-M values were very low in nonPORD subjects, and 11-DOF-M and 11-OHΔ4A-M (5β-reduced) values were very low in all subjects examined, so that, in conjunction with the interference of unknown steroids probably of fetal adrenal origin, it was impossible to measure them precisely.

Upper panel: Simplified schematic representation indicating impaired cholesterologenesis and steroidogenesis in POR mutations. DOC: deoxycorticosterone; 17-OHP: 17α-hydroxyprogesterone; 11-DOF: 11-deoxycortisol; 21-DOF: 21-deoxycortisol; DHEA: dehydroepiandrosterone; Δ4A: androstenedione; 11-OHΔ4A: 11-hydroxyandrostenedione; T: testosterone; and DHT: dihydrotestosterone. POR dependent CYP51A1 (lanosterol 14α-demethylase), CYP17A1 (17α-hydroxylase and 17,20 lyase), CYP21A2 (21-hydroxylase), and CYP19A1 (aromatase) are impaired in patients with POR mutations. The urine metabolites of progesterone, 17-OHP, 21-DOF, DHEA, Δ4A, and 11-OHΔ4A are informative for the diagnosis of PORD (shown in red), whereas those of 17-OH pregnenolone, aldosterone, and cortisol are nondiagnostic (shown in blue). The urine metabolites of pregnenolone, DOC, corticosterone, 11-DOF are not measured precisely because of the low values and the interference of probably fetal adrenocortex derived unknown steroids. The “backdoor” pathway in which 17-OHP is sequentially converted into DHT via 5α-pregnane-3α,17α-diol-20-one and androsterone is indicated by dotted lines with arrows. Lower panel: Representative results of urine steroid profile analysis. M(s): metabolite(s); PD: pregnanediol; PT: pregnanetriol; Ptl: pregnanetriolone; Σ: the sum of DHEA, androstenediol, 16α-hydroxyDHEA, 16β-hydroxyDHEA, 16-oxoandrostenediol, and androstenetriol; An: androsterone; Et: etiocholanolone; and 11-OHAn: 11-hydroxyandorosterone. The data in cases 1–3 are primarily consistent with combined CYP21A2 and CYP17A1 deficiency and permit the differentiation from transient elevation of serum 17-OHP (Transient), classical 21-hydroxylase deficiency (21-OHD), and nonclassical 21-hydroxylase deficiency (NC21-OHD).
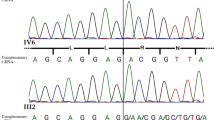
Mutation analysis of POR. Cases one through three were homozygous for R457H at exon 11. This mutation should create a BsaXI restriction site, and this was confirmed by the BsaXI digestion. The parents of cases two and three were heterozygous for R457H.
DISCUSSION
POR mutation analysis revealed homozygosity for R457H in cases one through three. This is consistent with R457H being the most frequent mutation in the Japanese patients (4). Indeed, R457H accounts for 17 of 30 mutant alleles reported in the Japanese patients (1,4,6,17), and homozygosity for R457H has been identified in four Japanese patients who were born to nonconsanguineous parents (4,6). Furthermore, a founder effect has been suggested for R457H in the Japanese patients on the basis of invariable association between R457H and a specific pattern of 10 single nucleotide polymorphisms in the POR gene (4), although R457H has also been identified in four non-Japanese patients (2,6).
Clinical features in cases one through three would primarily be compatible with R457H retaining some residual activity (1,2,6) Since phenotypic severity is known to roughly correlate with residual POR activity (6), this would explain the absent or mild nondiagnostic skeletal lesion in cases one through three, the lack of adrenal crisis in cases one through three, and the normally masculinized external genitalia in case one. Although cases two and three had severely virilized external genitalia, this would be explained by two factors. First, since the placenta is known to produce a large amount of estrogens that are converted from androgens by CYP19A1 (18), impaired placental CYP19A1 activity would result in a production of a fairly large amount of T and Δ4A in the presence of the residual POR activity, leading to the virilized genitalia. Such impaired CYP19A1 activity would also be relevant to maternal virilization and low urine E3 excretion during pregnancy (5,8,9). Second, since 17-OHP should be accumulated in PORD, this would drive the sequential conversion of 17-OHP into DHT via androsterone (2,19) (Fig. 2, upper part). Although such a T independent “backdoor” pathway to DHT production is mediated by CYP17A1 as well as 5α-reductase and other enzymes (2,19). 5α-pregnane-3α,17α-diol-20-one is known to have a much higher affinity for CYP17A1 than 17-OHP (20), so that this pathway would function better than the conventional pathway in PORD. The “backdoor” pathway has been shown in the tammar wallaby pouch young testis and immature mouse testis, and implicated in the virilization of female patients with 21-OHD and PORD (19). In this regard, both factors would have contributed to the normal genitalia in case one.
Cases one through three were initially regarded as having nonclassical 21-OHD and/or aromatase deficiency but were subsequently demonstrated to have PORD. In this context, the present and the previous studies (1,2,4,5) imply that comprehensive assessment of several key findings is essential for the precise diagnosis of PORD. They include: 1) maternal virilization and low E3 during the pregnancy; 2) the absence of skin pigmentation and the presence of subtle skeletal features in the physical examination; 3) the combined CYP21A2 and CYP17A1 deficiency in the adrenal function studies; and 4) the primary hypogonadism and PCO in the gonadal assessment. In particular, urine steroid profile analysis, though not prevailed, appears to be highly useful, because it invariably shows the combined enzymatic defect characteristic of PORD (2,4,21, this report). Thus, urine steroid profile analysis should serve for a screening of PORD in patients with apparent 21-OHD or aromatase deficiency, as well as in those with ABS-like skeletal phenotype as the primary phenotype.
For the urine steroid profile analysis, several matters should be pointed out. First, despite the impaired activity of CYP17A1, urine androsterone accounting for the vast majority of Δ4A-Ms tended to be increased, especially before four months of age. Since androsterone is produced in the “backdoor” pathway that could exist during the first several months of life (2) (Fig. 2, upper part), androsterone derived from the “backdoor” pathway may have been detected in the urine. In this context, the results imply that such androsterone is unlikely to undergo the conversion into 11-hydroxyandrosterone and etiocholanolone that were grossly normal or low in cases one through three. Thus, the DHEA-M and the 11-OHΔ4A-M (5α-reduced) values may represent the in vivo CYP17A1 activity. Second, the ratio between 11-OHΔ4A-M (5α-reduced) and progesterone-M appears to serve as an excellent indication to discriminate between 21-OHD and PORD. Third, Shackleton et al. (22) have suggested that urine pregnadienol (the artifact of pregnenediol disulfate detectable exclusively in PORD) can be viewed as a hallmark analyte, although it was not measured in this study. Lastly, Shackleton et al. (21) have also reported that steroid profile analysis of the maternal urine can detect the “backdoor” pathway derived steroids including androsterone, thereby permitting the prenatal diagnosis of PORD.
In summary, the results indicate that PORD can manifest clinical features similar to those in 21-OHD and in aromatase deficiency. In this context, it is possible that the previous studies on PORD may have primarily focused on patients with relatively severe manifestations such as ABS-like skeletal features. In addition, POR mutations may underlie in patients with clinical diagnosis of 21-OHD in whom CYP21A2 mutations have not been identified (23). Further studies will permit a better definition of the phenotypic spectrum of PORD.
Abbreviations
- ABS:
-
Antley-Bixler syndrome
- Δ4A:
-
androstenedione
- DHEA:
-
dehydroepiandrosterone
- DOF:
-
deoxycortisol
- E3:
-
estriol
- M(s):
-
metabolite(s)
- PCO:
-
polycystic ovary
- POR:
-
cytochrome P450 oxidoreductase
- PORD:
-
cytochrome P450 oxidoreductase deficiency
- T:
-
testosterone
- 17-OHP:
-
17α-hydroxyprogesterone
- 21-OHD:
-
21-hydroxylase deficiency
References
Flück CE, Tajima T, Pandey AV, Arlt W, Okuhara K, Verge CF, Jabs EW, Mendonca BB, Fujieda K, Miller WL 2004 Mutant P450 oxidoreductase causes disordered steroidogenesis with and without Antley-Bixler syndrome. Nat Genet 36: 228–230
Arlt W, Walker EA, Draper N, Ivison HE, Ride JP, Hammer F, Chalder SM, Borucka-Mankiewicz M, Hauffa BP, Malunowicz EM, Stewart PM, Shackleton CH 2004 Congenital adrenal hyperplasia caused by mutant P450 oxidoreductase and human androgen synthesis: analytical study. Lancet 363: 2128–2135
Miller WL 2004 P450 oxidoreductase deficiency: a new disorder of steroidogenesis with multiple clinical manifestations. Trends Endocrinol Metab 15: 311–315
Fukami M, Horikawa R, Nagai T, Tanaka T, Naiki Y, Sato N, Okuyama T, Nakai H, Soneda S, Tachibana K, Matsuo N, Sato S, Homma K, Nishimura G, Hasegawa T, Ogata T 2005 Cytochrome P450 oxidoreductase gene mutations and Antley-Bixler syndrome with abnormal genitalia and/or impaired steroidogenesis: molecular and clinical studies in 10 patients. J Clin Endocrinol Metab 90: 414–426
Cragun DL, Trumpy SK, Shackleton CH, Kelley RI, Leslie ND, Mulrooney NP, Hopkin RJ 2004 Undetectable maternal serum uE3 and postnatal abnormal sterol and steroid metabolism in Antley-Bixler syndrome. Am J Med Genet A 129: 1–7
Huang N, Pandey AV, Agrawal V, Reardon W, Lapunzina PD, Mowat D, Jabs EW, Van Vliet G, Sack J, Fluck CE, Miller WL 2005 Diversity and function of mutations in p450 oxidoreductase in patients with Antley-Bixler syndrome and disordered steroidogenesis. Am J Hum Genet 76: 729–749
Stewart PM 2002 The adrenal cortex. In: Larsen PR, Kronenberg HM, Melmed S, Polonsky KS (eds) Williams Textbook of Endocrinology. 10th ed. Saunders, Philadelphia 491–551 pp
Shozu M, Akasofu K, Harada T, Kubota Y 1991 A new cause of female pseudohermaphroditism: placental aromatase deficiency. J Clin Endocrinol Metab 72: 560–566
Conte FA, Grumbach MM, Ito Y, Fisher CR, Simpson ER 1994 A syndrome of female pseudohermaphrodism, hypergonadotropic hypogonadism, and multicystic ovaries associated with missense mutations in the gene encoding aromatase (P450arom). J Clin Endocrinol Metab 78: 1287–1292
Miller WL 2001 The adrenal cortex and its disorders. In: Brook CGD, Hindmarsh PC, Jacobs HS (eds) Clinical Pediatric Endocrinology. 4th ed. Blackwell Science, Oxford pp 321–376
Lashansky G, Saenger P, Fishman K, Gautier T, Mayes D, Berg G, Di Martino-Nardi J, Reiter E 1991 Normative data for adrenal steroidogenesis in a healthy pediatric population: age- and sex-related changes after adrenocorticotropin stimulation. J Clin Endocrinol Metab 73: 674–686
Lashansky G, Saenger P, Dimartino-Nardi J, Gautier T, Mayes D, Berg G, Reiter E 1992 Normative data for the steroidogenic response of mineralocorticoids and their precursors to adrenocorticotropin in a healthy pediatric population. J Clin Endocrinol Metab 75: 1491–1496
Hasegawa Y 2003 Normal range of GnRH stimulation test. In: Hasegawa Y (ed) Let's enjoy pediatric endocrinology. 3rd ed. Sindan-to-tiryou-sha, Tokyo, Japan pp 260–262
Kreutzmann DJ, Cowell CT, Howard NJ, De Souza M, Silink M 1989 Congenital adrenal hyperplasia family studies using the short ACTH test. Aust Paediatr J 25: 340–345
Homma K, Hasegawa T, Masumoto M, Takeshita E, Watanabe K, Chiba H, Kurosawa T, Takahashi T, Matsuo N 2003 Reference values for urinary steroids in Japanese newborn infants: gas chromatography/mass spectrometry in selected ion monitoring. Endocr J 50: 783–792
New MI, Lorenzen F, Lerner AJ, Kohn B, Oberfield SE, Pollack MS, Dupont B, Stoner E, Levy DJ, Pang S, Levine LS 1983 Genotyping steroid 21-hydroxylase deficiency: hormonal reference data. J Clin Endocrinol Metab 57: 320–326
Adachi M, Tachibana K, Asakura Y, Yamamoto T, Hanaki K, Oka A 2004 Compound heterozygous mutations of cytochrome P450 oxidoreductase gene (POR) in two patients with Antley-Bixler syndrome. Am J Med Genet 128: 333–339.
Grumbach MM, Hughes IA, Conte FA 2002 Disorders of sex differentiation. In: Larsen PR, Kronenberg HM, Melmed S, Polonsky KS (eds) Williams textbook of endocrinology. 10th ed. Saunders, Philadelphia pp 842–1002
Auchus RJ 2004 The backdoor pathway to dihydrotestosterone. Trends Endocrinol Metab 15: 432–438
Wilson JD, Auchus RJ, Leihy MW, Guryev OL, Estabrook RW, Osborn SM, Shaw G, Renfree MB 2003 5alpha-androstane-3alpha, 17beta-diol is formed in tammar wallaby pouch young testes by a pathway involving 5alpha-pregnane-3alpha, 17alpha-diol-20-one as a key intermediate. Endocrinology 144: 575–580
Shackleton C, Marcos J, Arlt W, Hauffa BP 2004 Prenatal diagnosis of P450 oxidoreductase deficiency (ORD): a disorder causing low pregnancy estriol, maternal and fetal virilization, and the Antley-Bixler syndrome phenotype. Am J Med Genet A 129: 105–112
Shackleton C, Marcos J, Malunowicz EM, Szarras-Czapnik M, Jira P, Taylor NF, Murphy N, Crushell E, Gottschalk M, Hauffa B, Cragun DL, Hopkin RJ, Adachi M, Arlt W 2004 Biochemical diagnosis of Antley-Bixler syndrome by steroid analysis. Am J Med Genet A 128: 223–231
Nimkarn S, Cerame BI, Wei JQ, Dumic M, Zunec R, Brkljacic L, Skrabic V, New MI, Wilson RC 1999 Congenital adrenal hyperplasia (21-hydroxylase deficiency) without demonstrable genetic mutations. J Clin Endocrinol Metab 84: 378–381
Author information
Authors and Affiliations
Corresponding author
Additional information
Supported by a grant for Child Health and Development from the Ministry of Health, Labor, and Welfare (17C-2), and by a Grant-in-Aid for Scientific Research on Priority Areas (16086215).
Rights and permissions
About this article
Cite this article
Fukami, M., Hasegawa, T., Horikawa, R. et al. Cytochrome P450 Oxidoreductase Deficiency in Three Patients Initially Regarded as Having 21-Hydroxylase Deficiency and/or Aromatase Deficiency: Diagnostic Value of Urine Steroid Hormone Analysis. Pediatr Res 59, 276–280 (2006). https://doi.org/10.1203/01.pdr.0000195825.31504.28
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/01.pdr.0000195825.31504.28
This article is cited by
-
Congenital adrenal hyperplasia, disorders of sex development, and infertility in patients with POR gene pathogenic variants: a systematic review of the literature
Journal of Endocrinological Investigation (2022)
-
Regulation of P450 oxidoreductase by gonadotropins in rat ovary and its effect on estrogen production
Reproductive Biology and Endocrinology (2008)
-
P450 oxidoreductase deficiency and Antley–Bixler syndrome
Reviews in Endocrine and Metabolic Disorders (2007)
