
Abstract
Variability in the expression of apoptotic and inflammatory mediators with time after an ischemic insult and their role in the expansion of cerebral infarcts are still controversial. This study examines DNA degradation and the expression of activated caspase-3 and iNOS, inducible nitric oxide (iNOS) in the human developing brain. Autopsy specimens from children with a neuropathologic diagnosis of focal ischemic infarct were included in the study. The specimens were classified based on the clinical history as acute (<24 h, n = 5), subacute (24–72 h, n = 8), or old (>72 h, n = 6) infarcts. Immunohistochemical staining for caspase-3, iNOS and TUNEL were then performed on all infarcts alongside age-matched controls. TUNEL staining was detected in regions of all infarcts. Expression of iNOS was significantly higher than that of caspase-3 in the penumbra of subacute infarcts (p = 0.02). Glial fibrillary acidic protein and iNOS staining co-localized in the penumbra of acute and subacute infarcts. These results suggest that cell death continues to occur for more than 3 d post ischemic insult. Cell death in the penumbra of subacute infarcts is partially caspase-3 independent and may be attributed to nitric oxide. Astrocytes are a source of iNOS and may play a role in the evolution of pediatric brain injury days after the initial insult.
Similar content being viewed by others
Main
Cerebrovascular disorders are among the top 10 causes of death in children, with rates being highest in the first year of life. Population-based studies have identified incidence rates of pediatric ischemic stroke of 3.3–13/100,000 (1–3). The incidence rate of neonatal stroke is approximately 1/4000 live births per year (4,5). Incidence and prevalence of stroke in children is likely to increase because of increased awareness and improvement in radiographic diagnosis. Although several studies have been published on the pathophysiology of infarcts in the adult brain, very little is known about the pathophysiology of pediatric infarcts.
Studies using animal models of neonatal stroke have shown that activated caspase-3 (6–8) and nitric oxide (9–11) are mediators of ischemic injury in the developing brain. Several studies (12–15) have shown that necrotic cell death predominated in the ischemic core whereas apoptosis occurred in the penumbra. A more recent study showed that neuronal caspase-3 was expressed in both the ischemic core and penumbra after transient focal ischemia with a more rapid increase in the penumbra (16). Nitric oxide synthase (NOS) activity, on the other hand, was shown to be higher in the core compared with the penumbra (9). Furthermore, mice lacking iNOS are less vulnerable to ischemic injury compared with wild type (10), suggesting iNOS blockade may serve to limit the extent of ischemic damage.
Glial cells are thought to be more resistant to injury in the CNS. However, astrocytic swelling is an early response to ischemic injury. Following focal ischemia, astrocytes will increase uptake of glutamate, K+ ions, and lactate in an attempt to reduce the accumulation of these molecules in the extracellular space. Consequently, astrocytes will swell leading to cerebral edema that will exacerbate the ischemic injury and cause death (17). In addition to trying to protect the homeostasis of the extracellular milieu, astrocytes maintain their ability to communicate with each other and with distant neurons through gap junctions that remain open during an ischemic injury (18). Consequently, proapoptotic substances such as iNOS, shown to be expressed in rat glial cells following ischemia (11,19,20), can diffuse to neighboring cells via this network and cause further damage.
The variability in the expression of cell death mediators with time after an ischemic insult and their role in the expansion of cerebral infarcts are still controversial, however (6–8). Moreover, attempts to extrapolate from animal models have limitations when it comes to clinical applications. The objective of this study was to examine the regional distribution and the time course of caspase-3- and iNOS-mediated injury following ischemia in the human developing brain using clinically defined acute, subacute and old infarcts. A better understanding of the role of these proteins in human pediatric ischemic infarcts may point toward useful therapeutic targets.
PATIENTS AND METHODS
Patient population.
The database in the Department of Pathology at the Hospital for Sick Children was searched for autopsy specimens from children registered from January 1990 to July 2004 with a neuropathologic diagnosis of cerebral ischemic infarct. Specimens were included in the study if 1) patient was born at term; 2) patient age <18 y; and 3) cerebral infarct had well-defined borders. Patients with global ischemia were excluded from the study. After reviewing the charts, infarcts were classified based on the clinical history as acute if the time between clinical neurologic compromise and autopsy was up to 24 h, as subacute if that time was 24–72 h, and old if more than 72 h. Age-matched brain tissue sections with normal histology were used as controls. The study was approved by the Research Ethics Board at our institution.
Neuropathological examination.
Paraffin-embedded sections of postmortem pediatric human brains stained with hematoxylin and eosin (H&E) were reviewed by the neuropathologist (C.H.) to confirm the diagnosis of focal ischemic infarct. The area of the infarct was defined by loss of normal H&E staining pattern of brain parenchyma with specific cellular changes depending on the age of the infarct. Acute infarcts showed neuronal eosinophilia with nuclear pyknosis and vacuolation of surrounding neuropil. In addition, neutrophil and early macrophage infiltration was seen in the subacute infarcts. Vascular proliferation, cystic cavity formation, abundant foamy histocytes, and reactive astrocytosis were characteristics of old infarcts. Brain parenchyma directly adjacent to the ischemic “core” was defined as the ischemic margin or “penumbra” (Fig. 1). None of the age-matched control sections showed any pathologic changes.

Hematoxylin and eosin (H&E) staining of subacute infarct in the pons of a 3-wk-old female who died of neonatal meningitis. Core of the infarct was defined by loss of normal H&E staining pattern of brain parenchyma with specific cellular changes depending on the age of the infarct. Normal brain parenchyma directly adjacent to the ischemic core (arrow) is the ischemic margin or “penumbra.”
Tissue preparation.
Five micrometers of formalin fixed, paraffin embedded tissue section were mounted on positive-charged microscope slides. Tissue sections were then baked overnight at 60°C, de-waxed in xylene, and hydrated to distilled water through decreasing concentrations of alcohol.
TUNEL assay.
DNA damaged cells were detected using the in situ end labeling assay for TUNEL (terminal deoxynucleotidyl transferase-mediated DNA nick-end labeling) adapted to an automated immuno/in situ hybridization technique (Discovery Ventana Medical Systems, Inc., Tucson, AZ). Before staining, the sections were blocked for endogenous peroxidase and a biotin block, with subsequent online digestion with protease I (Ventana Medical Systems) for 12 min. The assay uses recombinant terminal deoxynucleotidyl transferase (Tdt) (Invitrogen, Carlsbad, CA) for adding homo-polymer tails to the 3′ ends of cleaved DNA, characteristic in cells undergoing programmed cell death. Biotin 16-dUTP (Roche Diagnostics, Basel, Switzerland) was the label used for this reaction. Colorimetric visualization using avidin-horse radish peroxidase and 3,3′-diaminobenzidine (DAB) detection method was performed. The counterstain was hematoxylin.
Activated caspase-3.
Antibody against cleaved caspase-3 (Cell Signaling Technology, Beverly, MA) was pretreated with a heat-induced epitope retrieval technique using citrate buffer at pH 6.0, and used at a dilution of 1/25. Immunohistochemical procedures were performed on the Ventana NEXES auto-immunostainer (Ventana Medical Systems). Immunodetection was performed using an ABC (avidin/biotin complex) system, employing the Ventana i-View DAB (3-3′- diaminobenzidine) Detection Kit. All tissue sections were treated for endogenous peroxidase and biotin. The counterstain of preference was hematoxylin, for nuclear detail.
iNOS staining.
The antibody against iNOS (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) required protease I (Ventana Medical Systems) pretreatment for 16 min as part of the automated staining protocol. The antibody was then used at a dilution of 1/40. The use of this antibody to detect iNOS is well documented in the literature (45,46).
TUNEL and neurofilament.
Antibody for neurofilament is a well-established marker for detecting neurons. Double staining with neurofilament and TUNEL was done to identify the type of the dying cells. TUNEL staining was carried out as described above. Following that, the immunohistochemical procedure for neurofilament (Sanbio, Am Uden, the Netherlands) was performed. Sections were retreated with endogenous peroxidase and biotin before incubation with neurofilament at 1/50 dilution for 60 min at room temperature. Immunodetection was performed employing the Elite ABC detection system (Vector Laboratories, Burlingame, CA), as described by the manufacturer, in conjunction with the Vector VIP substrate kit (Vector Laboratories), as the chromogen, producing a purple reaction product. The counterstain of preference was hematoxylin, for blue nuclear detail.
GFAP and iNOS.
Antibodies to GFAP are routinely used to identify glial cells particularly astrocytes. The morphology of iNOS-positive cells suggests that these cells are astrocytes. To confirm the identity of iNOS-positive cells by immunocytochemistry, we co-stained them with antibodies against iNOS and GFAP. Sections were digested with 0.5% pepsin at 37°C for 30 min, and blocked with biotin and goat normal serum before the application of the primary antibody. The incubation of anti-human GFAP (DAKO, Carpinteria, CA) at 1:1000 dilution was carried out at room temperature for 1 h, followed by anti-rabbit IgG secondary antibody and fluorescent-streptavidin (Vector Laboratories, Inc.). Anti-iNOS antibody (Santa Cruz Biotechnology) at 1:10 dilution was added to the slides and incubated at 4°C overnight. Then, the anti-mouse IgG secondary antibody and TRITC- streptavidin (Vector Laboratories Inc.) was applied to the slides and incubated at room temperature for 30 min. The slides were washed with 1× PBS and cover-slipped.
Cell counting.
Cell counting was performed independently by two investigators blinded to the clinical classification of infarcts (R.A. and C.H.) in the core and margin of each infarct. Averages of four to five visual fields were counted for each region at 40× magnification. The same areas were counted in the age-matched control brains for each immunostain. The numbers of positive cells were reported as mean ± SD.
Statistical Analysis.
The means ± SD of positive cells counted for different stains in the core and margin of each infarct were compared among acute, subacute and old infarcts. Statistically significant differences were analyzed using one-way analysis of variance.
RESULTS
A total of 19 patients registered in the database of the Department of Pathology in our institution from January 1990 to July 2004 with a neuropathological diagnosis of cerebral ischemic infarct met the inclusion criteria of our study. Of these 19 patients, 5 had acute infarcts, 8 had subacute infarcts, and 6 had old infarcts. None of these patients died of their ischemic infarcts. The clinical data on patients and controls (n = 5) are shown in Table 1.
DNA damage.
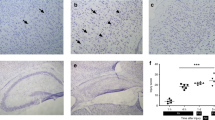
DNA damage, as detected in tissue sections using TUNEL immunostaining was significantly increased compared with control in the core and penumbra of all infarcts at all ages (representative sections are shown in Fig. 2). There was no statistically significant difference in the number of TUNEL-positive cells between the core and penumbra in all infarct ages (Table 2). Only rare TUNEL-positive cells were seen in the control sections (26 ± 12) and in the nonaffected areas outside the penumbra. TUNEL and neurofilament staining were detected in the same cells of all infarcts (Fig. 3), confirming the damaged cells to be neurons.

TUNEL staining in core (A,C,E) and penumbra (B,D,F) of acute (A–B), subacute (C,D), and old (E,F) infarcts in the hippocampus, left temporal lobe, and right frontal cortex, respectively.

TUNEL-positive cells are neurons. TUNEL staining and neurofilament immunostaining were detected in the same cell (arrow).
Caspase-3 activity.
There were large numbers of caspase-3-positive cells in the core and penumbra of acute, subacute, and old infarcts (Fig. 4). However, the number of caspase-3-positive cells significantly decreased in the penumbra of subacute compared with acute infarcts (Table 2). The number of caspase-3-positive cells was significantly less than the number of TUNEL-positive cells (Table 2) in the core (p < 0.0001) and penumbra (p = 0.01) of subacute infarcts. No significant difference between the two stains was found in the acute and old infarcts. Rare Caspase-3-positive cells were seen in the control sections (3 ± 6) and in the nonaffected areas outside the penumbra.

Activated caspase-3 detected by immunohistochemistry in core (A,C,E) and penumbra (B,D,F) of acute (A,B), subacute (C,D), and old (E,F) infarcts in the hippocampus, left temporal lobe, and left calcarine cortex, respectively.
iNOS staining.
There were large numbers of iNOS-positive cells in the core and penumbra of all infarcts at all ages. Very rare iNOS-positive cells were observed in controls (2 ± 2) and in the nonaffected areas outside the penumbra. There was a significant decrease in the number of iNOS-positive cells (Fig. 5) in the core (p < 0.05) but not the penumbra of subacute compared with acute infarcts. The number of iNOS-positive cells was significantly higher (p = 0.02) in the penumbra versus the core of subacute infarcts (Table 2). Comparing caspase-3 to iNOS staining, the number of caspase-3-positive cells was significantly higher in the core (p = 0.03), whereas iNOS-positive cells were higher in the penumbra (p = 0.04) of subacute infarcts. TUNEL staining, on the other hand, was significantly higher (p < 0.0001) when compared with iNOS staining in the core but not in the penumbra (p = 0.2) of subacute infarcts. These differences were not observed in the core and penumbra of acute and old infarcts (Table 2).

iNOS immunohistochemical staining in core (A,C,E) and penumbra (B,D,F) of acute (A,B), subacute (C,D), and old (E,F) infarcts in the pons, left temporal lobe, and left calcarine cortex, respectively.
GFAP and iNOS staining.
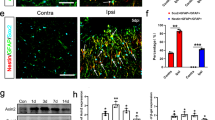
Many iNOS-positive cells were also positive for GFAP staining in the penumbra of acute and subacute infarcts (Fig. 6). None of the control sections stained with GFAP had iNOS-positive cells. These results indicate that the iNOS is being produced by astrocytes in the penumbra up to 72 h post ischemic infarct.

Localization of iNOS in astrocytes. Penumbra of acute (A,C,E) and subacute (B,D,F) were stained with GFAP antibody (A,B) and iNOS MAb (C,D) and were visualized with FITC-streptavidin (green) and TRITC- streptavidin (red), respectively. GFAP-positive cells were co-stained with iNOS as shown by merging the two images in E and F (yellow).
DISCUSSION
This is one of very few studies to look at the mechanism of injury in the human developing brain after an ischemic insult. We report four important findings: 1) cell death continues to occur in neurons in the core and penumbra of stroke for more than 72 h; 2) prolonged activation of caspase-3 in core and penumbra beyond 72 h; 3) iNOS prolonged expression in the core and penumbra of the infarct for more than 72 h; and 4) iNOS is expressed in astrocytes following ischemic injury.
TUNEL staining remained high in acute, subacute, and old infarcts, showing that cell death continued to occur in the core and penumbra of stroke for more than 72 h. This is consistent with what Pulera et al. (7) and Zhu et al. (8) have shown where TUNEL positivity was detected 72 h and 48 h, respectively, following ischemic insult in neonatal rats. However, in adult rat brain the peak of TUNEL immunostaining was between 12 and 24 h following focal ischemia (21).
We found that activated caspase-3 continued to be detected for more than 72 h after the ischemic insult. Our findings are supported by the work of others. Pulera et al. (7) reported the detection of apoptosis at 1, 3, and 4 d after ischemic insult in a neonatal stroke model using two antibodies specific for activated caspase-3. More recently, caspase-3 reactivity was reported at 24 and 72 h (7,8) after ischemic injury in a neonatal animal stroke model and remained high in some regions of the brain up to 7 d after the insult (8). In adult mouse brain, activated caspase-3 was detected 12–24 h after reperfusion (22). These data, taken together, suggest that cell death in the developing brain continues for longer periods of time compared with mature brain after an ischemic insult.
In the subacute infarcts, the number of cells staining with caspase-3 is less than that staining with TUNEL suggesting both caspase-3-dependent and -independent cell death. Caspase-3 and TUNEL reactivity are not significantly different in the acute and old infarcts, suggesting that cell death is primarily caspase-3 dependent at these stages. In contrast, Zhu et al. (8) reported that caspase-3 positivity exceeded TUNEL positivity at 3 h post ischemia, but the two stains completely co-localized at 24 h in a neonatal stroke model.
The above results taken together suggest that cell death in the developing brain is ongoing for at least 72 h following an ischemic insult. A similar prolonged cell death has not been shown in adult brain yet. Caspase-3-dependent cell death may predominate acutely (first 24 h) but in subacute infarcts caspase-3-independent pathways start playing a role in mediating cell death.
A potential weakness of this study, necessitated by the scarcity of human tissue, is the broad age range of included patients (1 wk to 16 y of age) as well as the differences in infarct location. To address this we examined our data to look for potential differences in immuno-positive cell counts related to patient age (<6 mo versus >6 mo) or infarct location (infra- versus supratentorial). There were no significant differences in our data in the number of TUNEL-positive or caspase-3- or iNOS-immuno-positive cells in the acute and subacute infarcts based on patient age or infarct location (data not shown). Nevertheless, as our study was not designed to address this question, we cannot exclude the possibility that the role of caspase-3-dependent and -independent pathways may vary as the brain matures within this pediatric population.
iNOS is expressed in the core and penumbra of the infarcted developing human brain for more than 72 h following the ischemic insult with a significant drop in iNOS expression after 24 h in the core but not the penumbra of subacute infarcts. Expression of iNOS in the penumbra of subacute infarcts is significantly higher than the expression of activated caspase-3, whereas the reverse is true in the infarct core. These results suggest that iNOS-mediates cell death in the penumbra in part, independently of caspase-3, while caspase-3-mediated cell death predominates in the core of subacute infarcts in the developing brain.
The fact that iNOS expression was still detected in the penumbra of infarcts older than 72 h is in agreement with studies showing that selective iNOS inhibition was neuroprotective 72 h post ischemia in the adult (23,24) and neonatal (25) rat hypoxic-ischemic model. In contrast, a decrease in NOS activity was reported by 24 h post ischemia in the rat pup (9). This reported early decrease in NOS activity may be attributed to neuronal NOS (nNOS) activity (26) and not iNOS.
Co-localization of iNOS and GFAP staining suggests that iNOS is expressed in astrocytes following ischemic injury in the developing brain. These results are consistent with previous reports showing iNOS expression by reactive astrocytes in adult rat (19) and neonatal rat cultured glial cells (20) following ischemia. However, in autopsy adult brains with neuropathological evidence of acute cerebral infarction, iNOS immunoreactivity was observed in neutrophils infiltrating the infarcted brain and in blood vessels within the infarcted territory (27). Expression of iNOS was shown predominately in neutrophils, and smooth muscle cells but not astrocytes in immature rats (28) and adult human brain (29) following traumatic brain injury. Our study provides evidence that astrocytes are a source of iNOS expression in the human developing brain following ischemia.
The physiologic role of astrocytic iNOS production following ischemia is not clear yet. Astrocytes are a major source of NO (30) but they also protect against oxidant damage (31–33). They contain the highest concentrations of antioxidants in the brain and provide neurons with extracellular precursors for neuronal glutathione (GSH) synthesis (33,34). Therefore, one possible mechanism of iNOS-induced ischemic injury is that increased expression of iNOS and subsequently NO may deplete cellular glutathione resulting in potentiation of ischemic injury as shown in adult (35–37) and neonatal (38) rat brain. Another possible mechanism of NO-induced ischemic injury is by disrupting astrocytic networks. It has been shown that NO uncouples gap junctions (39), which play an important role in neuronal survival following an ischemic injury (17,40). Elevation of neuronal Ca2+ induced by oxidative injury was larger with uncoupling treatment (41), suggesting that astrocytic gap junctions were also involved in neuronal intracellular Ca2+ homeostasis. However, nonselective nitric oxide synthase inhibitor did not alter the enhancement of oxidative neuronal injury by uncoupling of gap junction in mixed cultures of neurons and astrocytes (42). The deleterious effects of inflammatory iNOS expression to the ischemic brain are well documented in the literature (for reviews, see Refs. 26,43,44). However, the role of astrocytic iNOS expression in the context of ischemic injury is not clear yet. Further work is required to elucidate astrocyte-neuron communication following ischemia in the developing brain.
In summary, cell death continues to occur in the core and penumbra of stroke for more than 72 h in the human developing brain. This ongoing cell death is mediated by both caspase-dependent and caspase-independent mechanisms. iNOS-mediated cell death is shown to predominate in the penumbra of subacute infarcts, implicating it as a possible therapeutic target with a good therapeutic window.
Abbreviations
- GFAP:
-
glial fibrillary acidic protein
- iNOS:
-
inducible nitric oxide
References
deVeber G 2003 Arterial ischemic stroke in infants and children: an overview of current approaches. Semin Thromb Hemost 29: 567–573
Giroud M, Lemesle M, Gouyon JB, Nivelon JL, Milan C, Dumas R 1995 Cerebrovascular disease in children under 16 years of age in the city of Dijon, France: a study of incidence and clinical features from 1985 to 1993. J Clin Epidemiol 48: 1343–1348
Lynch JK, Hirtz DG, DeVeber G, Nelson KB 2002 Report of the National Institute of Neurological Disorders and Stroke Workshop on Perinatal and Childhood Stroke. Pediatrics 109: 116–123
Estan J, Hope P 1997 Unilateral neonatal cerebral infarction in full term infants. Arch Dis Child Fetal Neonatal Ed 76: F88–F93
Perlman JM, Rollins NK, Evans D 1994 Neonatal stroke: clinical characteristics and cerebral blood flow velocity measurements. Pediatr Neurol 11: 281–284
Nakajima W, Ishida A, Lange MS, Gabrielson KL, Wilson MA, Martin LJ, Blue ME, Johnston MV 2000 Apoptosis has a prolonged role in the neurodegeneration after hypoxic ischemia in the newborn rat. J Neurosci 20: 7994–8004
Pulera MR, Adams LM, Liu H, Santos DG, Nishimura RN, Yang F, Cole GM, Wasterlain CG 1998 Apoptosis in a neonatal rat model of cerebral hypoxia-ischemia. Stroke 29: 2622–2630
Zhu C, Wang X, Hagberg H, Blomgren K 2000 Correlation between caspase-3 activation and three different markers of DNA damage in neonatal cerebral hypoxia-ischemia. J Neurochem 75: 819–829
Ashwal S, Tone B, Tian HR, Cole DJ, Liwnicz BH, Pearce WJ 1999 Core and penumbral nitric oxide synthase activity during cerebral ischemia and reperfusion in the rat pup. Pediatr Res 46: 390–400
Nagayama M, Aber T, Nagayama T, Ross ME, Iadecola C 1999 Age-dependent increase in ischemic brain injury in wild-type mice and in mice lacking the inducible nitric oxide synthase Gene. J Cereb Blood Flow Metab 19: 661–666
You Y, Kaur C 2000 Expression of induced nitric oxide synthase in amoeboid microglia in postnatal rats following an exposure to hypoxia. Neurosci Lett 279: 101–104
Charriaut-Marlangue C, Margaill I, Represa A, Popovici T, Plotkine M, Ben Ari Y 1996 Apoptosis and necrosis after reversible focal ischemia: an in situ DNA fragmentation analysis. J Cereb Blood Flow Metab 16: 186–194
Chopp M, Li Y 1996 Apoptosis in focal cerebral ischemia. Acta Neurochir Suppl 66: 21–26
Li Y, Powers C, Jiang N, Chopp M 1998 Intact, injured, necrotic and apoptotic cells after focal cerebral ischemia in the rat. J Neurol Sci 156: 119–132
Towfighi J, Zec N, Yager J, Housman C, Vannucci RC 1995 Temporal evolution of neuropathologic changes in an immature rat model of cerebral hypoxia: a light microscopic study. Acta Neuropathol (Berl) 90: 375–386
Manabat C, Han BH, Wendland M, Derugin N, Fox CK, Choi J, Holtzman DM, Ferriero DM, Vexler ZS 2003 Reperfusion differentially induces caspase-3 activation in ischemic core and penumbra after stroke in immature brain. Stroke 34: 207–213
Anderson MF, Blomstrand F, Blomstrand C, Eriksson PS, Nilsson M 2003 Astrocytes and stroke: networking for survival?. Neurochem Res 28: 293–305
Cotrina ML, Kang J, Lin JH, Bueno E, Hansen TW, He L, Liu Y, Nedergaard M 1998 Astrocytic gap junctions remain open during ischemic conditions. J Neurosci 18: 2520–2537
Endoh M, Maiese K, Wagner J 1994 Expression of the inducible form of nitric oxide synthase by reactive astrocytes after transient global ischemia. Brain Res 651: 92–100
Kawase M, Kinouchi H, Kato I, Akabane A, Kondo T, Arai S, Fujimura M, Okamoto H, Yoshimoto T 1996 Inducible nitric oxide synthase following hypoxia in rat cultured glial cells. Brain Res 738: 319–322
Krupinski J, Lopez E, Marti E, Ferrer I 2000 Expression of caspases and their substrates in the rat model of focal cerebral ischemia. Neurobiol Dis 7: 332–342
Namura S, Zhu J, Fink K, Endres M, Srinivasan A, Tomaselli KJ, Yuan J, Moskowitz MA 1998 Activation and cleavage of caspase-3 in apoptosis induced by experimental cerebral ischemia. J Neurosci 18: 3659–3668
Cash D, Beech JS, Rayne RC, Bath PM, Meldrum BS, Williams SC 2001 Neuroprotective effect of aminoguanidine on transient focal ischaemia in the rat brain. Brain Res 905: 91–103
Tsuji M, Higuchi Y, Shiraishi K, Kume T, Akaike A, Hattori H 2000 Protective effect of aminoguanidine on hypoxic-ischemic brain damage and temporal profile of brain nitric oxide in neonatal rat. Pediatr Res 47: 79–83
Zhang F, Iadecola C 1998 Temporal characteristics of the protective effect of aminoguanidine on cerebral ischemic damage. Brain Res 802: 104–110
Iadecola C 1997 Bright and dark sides of nitric oxide in ischemic brain injury. Trends Neurosci 20: 132–139
Forster C, Clark HB, Ross ME, Iadecola C 1999 Inducible nitric oxide synthase expression in human cerebral infarcts. Acta Neuropathol (Berl) 97: 215–220
Clark RS, Kochanek PM, Schwarz MA, Schiding JK, Turner DS, Chen M, Carlos TM, Watkins SC 1996 Inducible nitric oxide synthase expression in cerebrovascular smooth muscle and neutrophils after traumatic brain injury in immature rats. Pediatr Res 39: 784–790
Orihara Y, Ikematsu K, Tsuda R, Nakasono I 2001 Induction of nitric oxide synthase by traumatic brain injury. Forensic Sci Int 123: 142–149
Heales SJ, Bolanos JP, Stewart VC, Brookes PS, Land JM, Clark JB 1999 Nitric oxide, mitochondria and neurological disease. Biochim Biophys Acta 1410: 215–228
Dringen R 2000 Metabolism and functions of glutathione in brain. Prog Neurobiol 62: 649–671
Peuchen S, Bolanos JP, Heales SJ, Almeida A, Duchen MR, Clark JB 1997 Interrelationships between astrocyte function, oxidative stress and antioxidant status within the central nervous system. Prog Neurobiol 52: 261–281
Wilson JX 1997 Antioxidant defense of the brain: a role for astrocytes. Can J Physiol Pharmacol 75: 1149–1163
Dringen R, Pfeiffer B, Hamprecht B 1999 Synthesis of the antioxidant glutathione in neurons: supply by astrocytes of CysGly as precursor for neuronal glutathione. J Neurosci 19: 562–569
Anderson MF, Sims NR 2002 The effects of focal ischemia and reperfusion on the glutathione content of mitochondria from rat brain subregions. J Neurochem 81: 541–549
Gotoh O, Yamamoto M, Tamura A, Sano K 1994 Effect of YM737, a new glutathione analogue, on ischemic brain edema. Acta Neurochir Suppl (Wien) 60: 318–320
Uemura Y, Miller JM, Matson WR, Beal MF 1991 Neurochemical analysis of focal ischemia in rats. Stroke 22: 1548–1553
Wallin C, Puka-Sundvall M, Hagberg H, Weber SG, Sandberg M 2000 Alterations in glutathione and amino acid concentrations after hypoxia-ischemia in the immature rat brain. Brain Res Dev Brain Res 125: 51–60
Bolanos JP, Medina JM 1996 Induction of nitric oxide synthase inhibits gap junction permeability in cultured rat astrocytes. J Neurochem 66: 2091–2099
Kirchhoff F, Dringen R, Giaume C 2001 Pathways of neuron-astrocyte interactions and their possible role in neuroprotection. Eur Arch Psychiatry Clin Neurosci 251: 159–169
Giaume C, Venance L 1998 Intercellular calcium signaling and gap junctional communication in astrocytes. Glia 24: 50–64
Blanc EM, Bruce-Keller AJ, Mattson MP 1998 Astrocytic gap junctional communication decreases neuronal vulnerability to oxidative stress-induced disruption of Ca2+ homeostasis and cell death. J Neurochem 70: 958–970
Bolanos JP, Almeida A 1999 Roles of nitric oxide in brain hypoxia-ischemia. Biochim Biophys Acta 1411: 415–436
Samdani AF, Dawson TM, Dawson VL 1997 Nitric oxide synthase in models of focal ischemia. Stroke 28: 1283–1288
Swindle EJ, Metcalfe DD, Coleman JW 2004 Rodent and human mast cells produce functionally significant intracellular reactive oxygen species but not nitric oxide. J Biol Chem 279: 48751–48759
Cardile V, Pavone A, Gulino R, Renis M, Scifo C, Perciavalle V 2003 Expression of brain-derived neurotrophic factor (BDNF) and inducible nitric oxide synthase (iNOS) in rat astrocytic cultures treated with Levetiracetam. Brain Res 976: 227–233
Acknowledgements
The authors thank Dr. Steven Ashwal and Dr. Jamie Hutchison for reviewing and discussing the manuscript and Dr. Conor Gallagher for technical help with confocal microscopy.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Askalan, R., Deveber, G., Ho, M. et al. Astrocytic-Inducible Nitric Oxide Synthase in the Ischemic Developing Human Brain. Pediatr Res 60, 687–692 (2006). https://doi.org/10.1203/01.pdr.0000246226.89215.a6
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/01.pdr.0000246226.89215.a6
This article is cited by
-
Hypoxia-induced expression of neuronal nitric oxide synthase in astrocytes of human corpus callosum
Brain Structure and Function (2021)
-
Modeling the effects of hyaluronic acid degradation on the regulation of human astrocyte phenotype using multicomponent interpenetrating polymer networks (mIPNs)
Scientific Reports (2020)
-
Role of XIAP gene overexpressed bone marrow mesenchymal stem cells in the treatment of cerebral injury in rats with cerebral palsy
Cancer Cell International (2019)
-
Aligned laminin core-polydioxanone/collagen shell fiber matrices effective for neuritogenesis
Scientific Reports (2018)
-
The anti-inflammatory role of extranuclear apurinic/apyrimidinic endonuclease 1/redox effector factor-1 in reactive astrocytes
Molecular Brain (2016)
