
Abstract
Isolated glycerol kinase deficiency (GKD) is an X-linked inborn error of metabolism that is either symptomatic or asymptomatic. GKD is due to deletions of, or mutations within, the GK gene, and there is no genotype-phenotype correlation. We identified three patients with asymptomatic GKD, determined that they had GK splice-site mutations, and studied the stability of their GK mRNA to understand the molecular mechanism of the GKD. All three patient mutations caused a frameshift and introduction of a premature stop codon. A fourth patient had an Alu insertion in intron 4 that led to alternative splicing. To study the effect of splice-site mutations on RNA species, we performed reverse transcriptase PCR and found only normal-sized products for all patients. Incubation with anisomycin to block nonsense-mediated decay (NMD), revealed two RNA species for each individual. Sequence analysis revealed that the larger bands represented the wild-type GK RNA and smaller bands represented mutant misspliced RNA, suggesting that the abnormal RNA species were targeted by NMD. Normal RNA species observed in each patient are likely responsible for their mild phenotypes. We speculate that influences on RNA processing and protein stability represent modifiers of the GKD phenotype.
Similar content being viewed by others

Main
Isolated GKD is an X-linked inborn error of metabolism (1). We have previously shown that GKD is due to deletions of, or mutations within, the glycerol kinase gene (GK) located on Xp21 that encodes a 524 aa protein (1). Isolated GKD is either asymptomatic or symptomatic with episodes of vomiting, acidosis (metabolic crises), and lethargy that can progress to coma or CNS crises. GKD is caused by large deletions, insertions, missense mutations, nonsense mutations, and splice-site mutations (1–3). Our previous work has shown that there is no genotype-phenotype correlation in GKD (2). We hypothesize that this lack of genotype-phenotype correlation in patients with GKD is due to the role of modifier genes in the GK phenotype (2,4).
NMD is a quality-control mechanism naturally occurring in cells to maintain homeostasis and destroy mRNA that contain premature stop codons (5,6). This process is evolutionarily conserved and is found in species from yeast (Saccharomyces cerevisiae) to worms (Caenorhabditis elegans) to humans (5). NMD occurs in the nucleus and cytoplasm (5,6), and evidence suggests that NMD and alternative splicing are coupled (7). Better understanding of this natural process to minimize the effect of DNA mutations in the genome will be important in understanding human disease.
The purpose of this investigation was to identify patients with GKD due to splice-site mutations and to determine the effect of the splice-site mutation on the GK mRNA stability. Our results indicate that GKD is one of the many disorders with splice-site mutations and that the aberrant transcripts are targeted for NMD.
METHODS
Individuals with isolated GKD.
Patient 020518-1 presented with pseudo-hypertriglyceridemia and asymptomatic GKD. He had four episodes of nephrolithiasis and glaucoma. Patient 101508-1 presented at 2 wk of age with hypertriglyceridemia. He had glyceroluria and normal plasma amino acids. He was born at 37 wk estimated gestational age by dates and 33 wk by exam. His Apgar scores were 9 and 9. Initial clinical laboratory results revealed hypoglycemia, elevated bilirubin, elevated lipids, elevated transaminases, thrombocytopenia, and megaloblastic anemia. FISH for Xp21 was normal. At a follow-up exam at 4 mo of age, he had normal growth parameters (5th to 25th percentile) and slightly decreased tone. Individual 120104-1 presented at 70 y of age with pseudo-hypertriglyceridemia and asymptomatic GKD. The final individual, 080112-1, with GKD had an Alu Y insertion (IVS4-52ins 316Alu) and has been previously described (8). He had pseudo-hypertriglyceridemia.
Establishment of cell lines and determination of GK mutations.
Patients with GKD were referred by outside physicians and informed consent using forms approved by the Institutional Review Boards of UCLA and Baylor College of Medicine was obtained. For each patient, blood samples were collected, a lymphoblastoid cell line (LBL) was established and maintained, and the GK Xp21 sequence including the 21 exons and intron-exon boundaries determined as previously described (8).
Cell culture and reverse transcriptase PCR analysis.
LBL cells were passaged 1–2 d before incubation with anisomycin (Sigma Chemical Co.-Aldrich, Milwaukee, WI) at a density of 2 ×106 cells per milliliter. On the day of the experiment, the cells were treated with anisomysin (Sigma Chemical Co.-Aldrich) for 2 h at a concentration of 100 μg/mL. Then, 2–3 ×107 cells were pelleted and total cellular RNA was isolated with a RNA Isolation Kit (Gentra Systems, Inc., Minneapolis, MN). Reverse transcriptase-PCR (RT-PCR) was performed for 40 cycles per manufacturer's instructions using the Titanium One-Step RT-PCR kit (BD Biosciences, San Jose, CA). RT-PCR products were sequenced at the UCLA Department of Human Genetics Sequencing and Genotyping Core Facility using an ABI 3700 sequencer (Applied Biosystems, Foster City, CA). The primers used for RT-PCR for patients 101508-1 and 080112-1 were 5′: CTATTGGTGTCAGCAAC 3′: and 5′: AACGGCCTTTTGAACTTTTC 3′:. For patient 020518-1 the primers were 5′: ACATCTGTACAGTGGACACC 3′: and 5′: CAGCTGAACTACTTAGTCATCATC 3′:. For patient 120104-1 the primers were 5′: AAAGCTGGGGCCTTGGAAGGT 3′: and 5′: GATTGTCTCTTAGCCAGCGAATAACAGCACCAG 3′:. RT-PCR products were run on a 2% agarose gel.
Glycerol kinase assays.
Glycerol kinase activity was determined using a radiochemical assay as previously described (9) in duplicate on three separate occasions and is expressed as percentage of normal control.
RESULTS
We identified three patients with GK splice-site mutations and studied their RNA stability (Table 1). The first (020518-1) had 6.8% of normal GK activity and a mutation, IVS3+ 1G>A. This resulted in deletion of exon 3 (107 bp), and a subsequent frameshift predicting a premature stop codon after 79 aa (Fig. 1, Table 1). The second patient (101508-1) had 8.3% of normal GK activity due to a mutation, IVS4-1G>A, which resulted in deletion of exon 5 (77 bp) and a frameshift predicting a premature stop codon after 119 aa (Fig. 1, Table 1). The third patient (120104-1) had 5.3% of normal GK activity due to a mutation IVS10+1G>T (Table 1). This resulted in deletion of exon 10 (43 bp) and a frameshift predicting a premature stop codon after 307 aa. The fourth patient was previously described (8). He had asymptomatic GKD and an AuY insertion in intron 4 (IVS4-52ins316Alu). His GK activity was 32% of normal. This AluY insertion lead to an alternative splice-site, deletion of exon 5 (77 bp) and a predicted a premature stop codon after 120 aa (8) (Table 1).

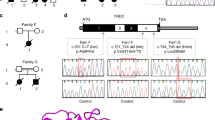
Schematic diagram of splice-site mutations in three GKD Patients. For each patient, the unique identifier (020518-1, 101508-1, and 120104-1) and the GK mutation (IVS3+1G>A, IVS4-1G>A, and IVS10+1G>T) are indicated. A schematic diagram of the genomic DNA of the patient in the region of the GK mutation is shown. E2 refers to exon 2. Exons are indicated as boxes and introns are indicated as lines. The primers used for RT-PCR are indicated (A-F, forward primer A; A-R, reverse primer A; B-F, forward primer B; B-R, reverse primer B). The sequences of the primers are indicated in “Methods.” The size of the normal (500 bp and 278 bp) as well as the misspliced (393 bp del E3, 424 bp del E5, and 235 bp del E10) RT-PCR products are indicated.
To study the effect of splice-site mutations on RNA species, we performed RT-PCR on RNA from each of the patients' LBL. In the absence of anisomycin, there were only normally sized products for all four patients (Fig. 2, lane 2, and Fig. 3, lanes 1–3). RT-PCR products of the appropriate size for the anticipated abnormally spliced forms were not observed. We hypothesized that abnormal RNA species were lost by NMD. To test this hypothesis, the patients' LBL were incubated in the presence of anisomycin to block NMD and two RNA species were seen for each patient (Fig. 2, lane 3, and Fig. 3, lanes 4–6). Sequence analysis revealed that for each patient the larger bands represented wild-type GK mRNA and smaller bands represented the predicted mutant misspliced RNA (data not shown).

Schematic diagram of splice-site mutations in a patient with GKD and an AluY insertion. (A) Schematic diagram of the genomic DNA in the region of the patient's GK mutation. Exons are indicated by boxes and introns by lines. The 316 bp AluY insertion is indicated and the arrow within it indicates the direction of the AluY sequence. (B) Agarose gel of the RT-PCR products. Lane 1, 1 kb DNA ladder; lane 2, RT-PCR product from patient 080112 LBL grown in the absence (–) of anisomycin; lane 3, RT-PCR product from patient 080112-1 LBL grown in the presence (+) of anisomycin; and lane 4, RT-PCR product from a normal control LBL grown in the presence (+) of anisomycin. The size of the RT-PCR products (245 bp and 168 bp) are indicated.

RT-PCR analysis of RNA from GKD patients LBL. Agarose gel of RT-PCR products from patient LBL (120104-1, lanes 1 and 4; 020518-1, lanes 2 and 5; 101508-1, lanes 3 and 6) in the presence (lanes 4–6) and absence (lanes 1–3) of anisomycin. RT-PCR product sizes (235 bp, 278 bp, 393 bp, 423 bp, and 500 bp) are indicated. Lane 7 is the 1 Kb DNA ladder.
DISCUSSION
We identified three patients with isolated GKD due to splice-site mutations and reexamined one patient with an AluY insertion leading to a splice-site alteration. Two other patients with splice-site mutations (IVS1+4A>G and IVS9-1G>T) within GK and associated with GKD have been described (3). The role of NMD in these patients was not determined.
RT-PCR analysis of our four patients' LBL revealed only the wild-type GK mRNA. This is surprising given that the mutation in three of these individuals affect completely conserved donor or acceptor splice sites and, therefore, one would expect very little normal splicing. However, the normal splicing seen in these patients may be due to the fact that other bases at the 5′ splice site are important and that the 3′: splice site has other important elements, including the branch site and polypyrimidine tract (10). The normally spliced wild-type GK seen in these patients is consistent with the residual GK activity and their asymptomatic course.
The mechanism of the Alu insertion causing a splice site mutation is unknown but sequence analysis of the alternatively spliced mRNA revealed loss of exon 5. Therefore, the Alu insertion may disrupt the 3′: splice site of intron 4, introduce new splicing regulatory elements, or interfere with existing splicing regulatory elements (10).
Inhibition of NMD revealed the alternatively spliced mRNA for each of these four individuals, indicating that the aberrant mRNA were targeted for destruction by NMD. Therefore, GKD is one of the increasing numbers of genetic diseases such as Marfan syndrome (11) and Netherton syndrome (12), in which the aberrant RNA generated by splice-site mutations are eliminated by NMD, and this phenomenon is important to understand their pathogenesis (13).
Interestingly, the four patients we describe here presented with the asymptomatic form of GKD, presumably because of varying levels of normal GK mRNA. The GK activity of these individuals ranged from 5.3% to 32% of normal. In addition, one of the patients with a splice-site mutation in GK (IVS1+ 4A>G) from the literature also had the asymptomatic form of GKD (3). Only one patient from the literature (IVS9-1G>T) had a history of metabolic and CNS crises indicating the symptomatic form of GKD (3). This suggests that these patients have GK activity in the indeterminant range (14) and that modifier genes are important in the phenotype of these individuals. We hypothesize that these modifier genes may be the genes encoding the proteins known to be involved in NMD, such as the SMG-1-like, RENT-1, hUPF1, RENT-2, hUPF2, and hUPF3A (5). In support of this hypothesis, Bitoun et al. (12) found varying levels of mutant SPINK5 mRNA on Northern blot analysis, suggesting “variable efficiency” NMD. In addition, other groups have noted that NMD is poorly understood, and there are intertissue and interindividual differences in NMD efficiency (13). Other modifiers that may be important in the variable phenotype of GKD and other Mendelian genetic disorders include the proteins that are important for RNA stability, protein translation, and protein folding (2,4).
Abbreviations
- aa:
-
amino acid
- GK:
-
glycerol kinase
- GKD:
-
glycerol kinase deficiency
- LBL:
-
lymphoblastoid cell line
- NMD:
-
nonsense-mediated decay
- RT–PCR:
-
reverse transcriptase PCR
References
McCabe E 2001 Disorders of glycerol metabolism. In: Scriver C, Beaudet A, Sly W, Valle D, Childs B, Vogelstein B (eds) Metabolic and Molecular Bases of Inherited Disease. McGraw-Hill, New York, pp 2217–2237
Dipple KM, Zhang YH, Huang BL, McCabe LL, Dallongeville J, Inokuchi T, Kimura M, Marx HJ, Roederer GO, Shih V, Yamaguchi S, Yoshida I, McCabe ER 2001 Glycerol kinase deficiency: evidence for complexity in a single gene disorder. Hum Genet 109: 55–62
Hellerud C, Adamowicz M, Jurkiewicz D, Taybert J, Kubalska J, Ciara E, Popowska E, Ellis JR, Lindstedt S, Pronicka E 2003 Clinical heterogeneity and molecular findings in five Polish patients with glycerol kinase deficiency: investigation of two splice site mutations with computerized splice junction analysis and Xp21 gene-specific mRNA analysis. Mol Genet Metab 79: 149–159
Dipple KM, McCabe ER 2000 Modifier genes convert “simple” Mendelian disorders to complex traits. Mol Genet Metab 71: 43–50
Byers PH 2002 Killing the messenger: new insights into nonsense-mediated mRNA decay. J Clin Invest 109: 3–6
Wilkinson MF, Shyu AB 2002 RNA surveillance by nuclear scanning?. Nat Cell Biol 4: E144–E147
Lewis BP, Green RE, Brenner SE 2003 Evidence for the widespread coupling of alternative splicing and nonsense-mediated mRNA decay in humans. Proc Natl Acad Sci U S A 100: 189–192
Zhang Y, Dipple KM, Vilain E, Huang BL, Finlayson G, Therrell BL, Worley K, Deininger P, McCabe ER 2000 AluY insertion (IVS4-52ins316alu) in the glycerol kinase gene from an individual with benign glycerol kinase deficiency. Hum Mutat 15: 316–323
Guggenheim MA, McCabe ER, Roig M, Goodman SI, Lum GM, Bullen WW, Ringel SP 1980 Glycerol kinase deficiency with neuromuscular, skeletal, and adrenal abnormalities. Ann Neurol 7: 441–449
Baralle D, Baralle M 2005 Splicing in action: assessing disease causing sequence changes. J Med Genet 42: 737–748
Caputi M, Kendzior RJ Jr, Beemon KL 2002 A nonsense mutation in the fibrillin-1 gene of a Marfan syndrome patient induces NMD and disrupts an exonic splicing enhancer. Genes Dev 16: 1754–1759
Bitoun E, Chavanas S, Irvine AD, Lonie L, Bodemer C, Paradisi M, Hamel-Teillac D, Ansai S, Mitsuhashi Y, Taieb A, de Prost Y, Zambruno G, Harper JI, Hovnanian A 2002 Netherton syndrome: disease expression and spectrum of SPINK5 mutations in 21 families. J Invest Dermatol 118: 352–361
Holbrook JA, Neu-Yilik G, Hentze MW, Kulozik AE 2004 Nonsense-mediated decay approaches the clinic. Nat Genet 36: 801–808
Dipple KM, McCabe ER 2000 Phenotypes of patients with “simple” Mendelian disorders are complex traits: thresholds, modifiers, and systems dynamics. Am J Hum Genet 66: 1729–1735
Acknowledgements
The authors thank the patients for participating in research and the physicians, including Dr. Helen Hobbs, University of Texas Southwestern Medical Center at Dallas, for referring the patients included in this study.
Author information
Authors and Affiliations
Corresponding author
Additional information
This work was supported by National Institutes of Health grants R01 HD22563, P30 HD34610, R01 HD39322, and K08 DK60055 and the UCLA Mental Retardation Research Center (P30 HD04612).
Rights and permissions
About this article
Cite this article
Zhang, YH., Huang, BL., Jialal, I. et al. Asymptomatic Isolated Human Glycerol Kinase Deficiency Associated with Splice-Site Mutations and Nonsense-Mediated Decay of Mutant RNA. Pediatr Res 59, 590–592 (2006). https://doi.org/10.1203/01.pdr.0000203092.83074.eb
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/01.pdr.0000203092.83074.eb
This article is cited by
-
Inborn errors of cytoplasmic triglyceride metabolism
Journal of Inherited Metabolic Disease (2015)
