
Abstract
Holt-Oram syndrome (HOS) is an autosomal dominant heart-hand syndrome characterized by congenital heart disease (CHD) and upper limb deformity, and caused by mutations in the TBX5 gene. To date, the sensitivity of TBX5 genetic testing for HOS has been unclear. We now report mutational analyses of a nongenetically selected population of 54 unrelated individuals who were consecutively referred to our center with a clinical diagnosis of HOS. TBX5 mutational analyses were performed in all individuals, and clinical histories and findings were reviewed for each patient without reference to the genotypes. Twenty-six percent of the complete cohort was shown to have mutations of the TBX5 gene. However, among those subjects for whom clinical review demonstrated that their presentations met strict diagnostic criteria for HOS, TBX5 mutations were identified in 74%. No mutations were identified in those subjects who did not meet these criteria. Thus, these studies validate our clinical diagnostic criteria for HOS including an absolute requirement for preaxial radial ray upper limb malformation. Accordingly, TBX5 genotyping has high sensitivity and specificity for HOS if stringent diagnostic criteria are used in assigning the clinical diagnosis.
Similar content being viewed by others


Main
HOS is the most common of the heart-hand syndromes. It segregates in an autosomal dominant fashion and is estimated to occur in at least 1/100,000 live births (1). HOS is characterized by upper limb anomalies involving the preaxial radial ray and CHD (2,3). Upper limb deformity may be bilateral but asymmetric or even unilateral. The most common forms of CHD associated with HOS are ASD, usually of the ostium secundum variety, and VSD, usually occurring in the muscular trabeculated septum. Cardiac conduction disease may also occur, regardless of the presence or absence of structural cardiac disease (3). Lower limb, postaxial upper limb, craniofacial, pulmonary, genitourinary, and gastrointestinal malformations and intellectual and sensory deficits are not features of HOS (2–5)
Previously, we and others (6,7) showed that mutations in the TBX5 gene cause HOS. TBX5 is a member of the evolutionarily conserved T-box family of transcription factor genes (6). Other members of the T-box family have also been implicated in human disease, including TBX3 in ulnar-mammary syndrome (UMS) (8), TBX4 in “small nail patella” syndrome (9), TBX22 in X-linked cleft palate with ankyloglossia (10), and TBX1 in CHD associated with DiGeorge/velocardiofacial syndrome (11).
Despite application of TBX5 mutational analyses to patients with HOS in several studies (6,7,12,13), the sensitivity of TBX5 genetic testing for HOS has remained unclear. Three studies (12–14) reported surprisingly low sensitivities ranging from 22 to 35%. Mutational analyses of the coding regions for genes that cause other monogenic disorders exhibit higher sensitivities, e.g. 85% for sequence analysis of LMX1B in individuals with “nail patella” syndrome (15).
Potential explanations for the low mutation detection rates in HOS have included technical limitations of typical mutational analysis protocols that would prevent detection of large deletions (16) and/or genetic heterogeneity of the disorder. Genetic heterogeneity of HOS has been controversial (17–19). Although linkage to the TBX5 chromosome 12q24.1 locus has been the rule, genetic heterogeneity has been proposed in three families (17–19). However, of these families, one exhibited polydactyly, which is not considered a feature of HOS, one had insufficient power to determine statistically significant linkage, and diagnoses in two had the unusual requirement of bilateral limb deformity.
We now report mutational analyses of a consecutive series of 54 unrelated individuals with a clinical diagnosis of HOS referred to our center for genetic testing. We show that although TBX5 mutational analyses may initially appear to be relatively insensitive, as previously suggested, the sensitivity is markedly increased when coupled with rigorous adherence to diagnostic criteria. Thus, genetic testing combined with careful clinical evaluation provides effective diagnosis of HOS in the majority of cases.
METHODS
Clinical evaluations.
All subjects provided informed consent for these studies in accordance with the institutional review board of the Weill Medical College of Cornell University. For each individual, medical records were reviewed for the subject and first-degree family members to determine history and findings suggesting congenital anomalies. Records included results from upper limb radiographs, echocardiograms, electrocardiograms, karyotypes, and physical examinations (Tables 1 and 2).
Molecular genetic analyses.
Genomic DNA was extracted from peripheral lymphocytes and amplified by PCR as previously described (20). dHPLC (WAVE, Transgenomic Inc., Omaha, NE) analysis was performed for exons 2–9 of TBX5. These exons include the complete coding region of the TBX5 gene. Any samples with abnormal dHPLC tracings were subjected to automated bidirectional sequence analysis on an ABI 3100 (Applied Biosystems, Foster City, CA) with Big Dye terminators to determine the specific TBX5 sequence variant. Primer sequences and dHPLC conditions are summarized in Table 3. At least 200 normal chromosomes were screened by dHPLC and/or automated sequencing for any TBX5 variant identified. TBX5 variants were defined as mutations if they altered the protein structure and were not seen in ≥200 normal chromosomes.
RESULTS
Mutational analyses.
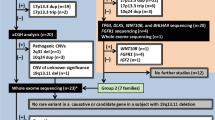
Fifty-four consecutive individuals with clinical diagnoses of HOS referred to our center were subjected to TBX5 mutational analysis. TBX5 mutations were observed in 14/54 (26%) of these individuals (Tables 1 and 2). One individual had two mutations (H170L; IVS5 + 1G>T), and two individuals shared the same mutation (Y136ter). Mutations included 6 nonsense, 1 deletion/frameshift, 5 splice site, and 3 missense mutations. Of these mutations, 11 had not been previously reported; 3 mutations (Y136ter, S196ter, and T223M) (12,14, 21) had been previously documented in patients with HOS. Six of the identified TBX5 mutations disrupt the T-box DNA binding domain, whereas the remainder occur downstream of the T-box and modify exons 7 and 8. No mutations of the alternatively spliced (22) exon 9 were observed.
Genotype-phenotype correlations.
Retrospective review of clinical records of all probands revealed features not consistent with a diagnosis of HOS in 35 of the 54 probands that had been referred for genetic testing. We had previously (3) defined clinical phenotypes diagnostic of HOS as the presence of preaxial radial ray malformation of at least one upper limb along with a personal or family history of septation defects (ASD, VSD) and/or atrioventricular conduction disease. In these 35 probands, atypical features include renal, lower limb, craniofacial, axillary and tracheal anomalies, deafness, polydactyly and isolated postaxial limb deformity. These are shown in Fig. 1 and are detailed in Table 1. Vertebral anomalies such as hemivertebrae were also observed in some of these individuals. Among these 35 individuals with clinical features inconsistent with HOS, no TBX5 mutations were found.

Upper limb malformation in individuals referred for TBX5 mutational analyses. (A) Typical upper limb malformation of HOS is evident in the left arm of subject #21. Note the absent thumb and malformation of the wrist causing disarticulation of the carpal bones with the radius bone. These defects are consistent with preaxial radial ray malformation. TBX5 mutational analyses of subject #21 genomic DNA revealed an IVS4 + 1G>T mutation. (B) The left hand of subject #16 is shown. The thumb and radial ray are normal. However, digits 4 and 5 are absent consistent with a postaxial malformation of the ulnar ray. TBX5 mutational analyses of subject #16 DNA were normal. However, the clinical presentation of subject #16 was more suggestive of ulnar-mammary syndrome than HOS. This diagnosis was confirmed by mutational analysis of TBX3 (performed as previously described (6)). A Val235ter TBX3 mutation in subject #16 as well as her mother, sister, son, and nephew all of whom exhibited similar postaxial upper limb deformity.
Nineteen of 54 patients had typical features of HOS (personal and/or family history of cardiac septation and/or conduction defects in the setting of preaxial radial ray deformity.) All 19 of these individuals had typical preaxial radial ray deformity without isolated postaxial upper limb or any lower limb malformation. Vertebral anomalies such as vertebral stenosis and vertebral fusions were also observed in three of these individuals, and given the report by Brassington et al. (12) of a TBX5 mutation in one HOS patient with typical limb and cardiac malformation as well as cervical vertebral fusions, such findings were not considered to exclude a diagnosis of HOS. TBX5 mutations were identified in 14/19 (74%) of these individuals with HOS. Family members with clinical presentations compatible with HOS, when available for testing, shared these mutations.
DISCUSSION
In this study, we observed TBX5 mutations in 26% of a cohort of individuals referred for HOS genetic testing. However, when this cohort was prescreened for individuals who strictly adhere to established HOS diagnostic criteria (personal and/or family history of cardiac septation and/or conduction defects in the setting of preaxial radial ray deformity), we found that our TBX5 dHPLC/sequencing based mutational analysis strategy, in fact, had a sensitivity of 74% in this population. No mutations were detected in individuals who had clinical features at variance with these HOS diagnostic criteria. Thus, we demonstrate that the addition of adherence to strict HOS diagnostic criteria to TBX5 mutational analysis provided markedly increased sensitivity in genetic testing for HOS. Moreover, given the absence of TBX5 mutations in a high-risk population of individuals but who had clinical features distinctive from HOS, we conclude that TBX5 mutational analysis is also 100% specific for HOS.
Individuals who did not meet HOS diagnostic criteria and who did not have identifiable TBX5 mutations often had clinical features that overlapped with HOS, e.g. hand malformations, cardiac septation defects, and/or atrioventricular conduction abnormalities. These features undoubtedly prompted the initial diagnosis of HOS. However, these individuals all had other notable clinical manifestations that were not typical of HOS. For instance, ulnar or lower limb involvement, renal anomalies, syndactyly involving digits other than the thumb, polydactyly, or craniofacial findings. Thus, observations of such features in individuals with upper limb deformity and/or cardiac septation defects should cast doubts upon any provisional diagnosis of HOS.
Importantly, a wide variety of heart-hand syndromes have been described (23,24) and although they may be familial as well, it is essential to recognize that they can be distinct from HOS. One subject (#16) with a personal and family history of ulnar ray abnormalities along with cardiac and renal malformations was subsequently shown to have a TBX3 mutation (Fig. 1) consistent with a diagnosis of UMS. Furthermore, several individuals in our series (e.g. subjects #4, #8, #18, #41, and #49) may have been more appropriately classified among the genetically heterogeneous category of VACTERL association, which is not necessarily inherited in a simple monogenic fashion. Thus, establishment of the correct clinical diagnosis can obviate the need for TBX5 mutational analysis or prompt other types of genetic testing and can have a significant impact on the genetic counseling of probands and their families.
Despite the high mutation detection rate achieved here among individuals who do meet clinical criteria for HOS, a TBX5 mutation was not identified in five such individuals. We were unable to test linkage to the TBX5 chromosome 12q24.1 locus in those cases meeting clinical criteria with no identifiable TBX5 mutation. This was either because the affected individual represented a sporadic case or the size of the individual's family was insufficient to provide statistically significant results. Although it is rare to detect mutations in all affected individuals with any monogenic disease, several factors may explain the less than 100% sensitivity observed in this series.
It is possible that the mutation detection methods used here would be unable to detect large TBX5 deletions that delete a complete exon. Because our analyses were limited to TBX5 coding regions (exons 2–9) and associated intron/exon boundaries, we would not detect mutations in the noncoding and/or regulatory regions of TBX5. These regions include at least three alternatively spliced versions of exon 1 that comprise a 5′ untranslated region that we have previously shown is critical for normal TBX5 expression (22).
Beyond the technical aspects of the mutational analyses performed, individuals without TBX5 mutations may also represent genetically distinct phenocopies of HOS, and four of the five HOS patients without TBX5 mutations had phenotypes and histories that would be unusual, albeit not impossible, for HOS patients. For instance, subject #10, who had preaxial radial ray anomalies as well as a VSD, also exhibited transposition of the great arteries and had a history of intrapartum infection and radiation exposure after having been conceived near Chernobyl in Russia. Subject #27, who also had preaxial radial ray anomalies, had a personal history of congenital structural heart malformation, an interatrial septal aneurysm, and a family history of idiopathic dilated cardiomyopathy. Neither isolated interatrial septal aneurysm nor idiopathic dilated cardiomyopathy has been associated with HOS, and both have been associated with genes and/or chromosomal loci distinct from TBX5 and chromosome 12q24.1 (25,26).
Notably, unique features of other syndromes that have partial overlap with HOS may exhibit incomplete and age-related penetrance. Thus, salient physical exam findings that ought to suggest a diagnosis other than HOS may not yet have been present at the time of examination of the patients in this series. For instance, poikiloderma and cataracts in the autosomal recessive radial ray malformation disorder Rothmund-Thomson syndrome (RTS) can present at advanced ages (27). Duane anomaly in Okihiro syndrome (also characterized by radial ray defects and congenital heart disease but caused by mutations in the SALL4 gene) exhibits incomplete penetrance and can be difficult to detect without focused ophthalmologic examination (28).
Along with several individuals who did not meet HOS diagnostic criteria for other reasons, three individuals (subjects #13, #36, and #39) who did meet those criteria also exhibited vertebral malformations suggestive of VACTERL association. A TBX5 mutation was identified in only one of these individuals (subject #36). Brassington et al. (12) also reported TBX5 mutations in one patient with HOS as well as vertebral anomalies, but the specific association with HOS remains unclear and TBX5 is not known to contribute to vertebral morphogenesis in the vertebrate embryo.
Some lessons can be gleaned from the TBX5 mutations that were identified in this cohort. Only two of the mutations (T233M and I106V) were missense; the remainder are predicted to produce TBX5 haploinsufficiency. These findings are consistent with the supposition that TBX5 haploinsufficient mutations are the most significant cause of HOS. Interestingly, despite suggestions from our previous population-based studies (22) that missense mutations at the 5′ end of the T-box more significantly affect the heart whereas those at the 3′ end of the T-box more significantly affect the upper limb, subject #52 with the 5′ T-box I106V mutation has upper limb malformation without evident cardiac disease, and subject #11 with the 3′ T-box T233M mutation has both an ASD and VSD as well as upper limb malformation. Similar inconsistencies have been reported by Brassington et al. (12), who also observed a T233M TBX5 mutation in two probands. Together with the findings here, these observations highlight the inability to apply statistical associations between genotypes and phenotypes seen in large populations to the individual proband. Furthermore, they emphasize the potential of any TBX5 mutation to affect development of both the heart and the limb in HOS.
However, our studies do support cautious interpretation of the pathogenesis of congenital heart defects when seen in families affected by HOS. The observation that subject #36 with HOS and an S196ter TBX5 mutation has tetralogy of Fallot (TOF) indicates the potential for HOS to present with conotruncal malformation. Given the absence of TBX5 protein in the developing human outflow tract (29), such congenital heart defects imply a potential secondary effect on the conotruncus when ventricular septation is impaired. On the other hand, other independent genetic mechanisms may be operant in such individuals with conotruncal malformation. Subjects #7 and #12 with HOS share the same Y136ter TBX5 mutation, and although both have preaxial radial ray malformation and cardiac conduction disease, only subject #12 and not #7 has a septation (atrial) defect. Evaluation of the family history for subject #7 revealed that a nephew without limb deformity did have TOF. However, when this individual was genotyped for the Y136ter TBX5 mutation, he was found to have no mutation. Thus, some other, as yet unidentified, genetic mechanism must exist in this family to produce conotruncal malformation. Similar questions are provoked by the clinical diagnosis of Long QT syndrome in subject #33 with typical HOS (upper limb deformity, ASD, and third-degree atrioventricular block) and an H271ter TBX5 mutation. Long QT syndrome has not previously been associated with HOS, but TBX5 has been now been shown in experimental animal models to be required for the normal establishment of the cardiac conduction system (30). To date, genetic testing of subject #33 has not revealed mutations at LQT1, LQT2, LQT3, LQT5, and LQT6 (Aftimos, unpublished data).
Although complex congenital heart disease has occasionally been associated with HOS and documented TBX5 mutations (12,14,22), this is not a common finding. In our series, several patients considered to have a diagnosis consistent with the strict clinical criteria for HOS had congenital heart malformation in addition to ASD, VSD, and/or conduction disease. However, TBX5 mutations were rarely detected in this group (subject #29 with aortic coarctation and subject #36 with TOF). Thus, it may still be appropriate to consider for TBX5 mutational analyses individuals who meet clinical criteria for HOS and who have congenital heart disease in addition to (but not instead of) septation defects and/or conduction disease.
In summary, we have shown that the application of strict clinical criteria for HOS is highly predictive for the identification of a TBX5 disease–causing mutation. The ability to confirm clinical diagnoses with genetic testing is a valuable asset in the management of affected individuals and their families. Informative genetic testing will be useful in identifying at-risk individuals, such as parents and siblings of those with HOS, and will permit counseling regarding specific recurrence risks for family members of an individual affected by HOS. Because of the phenotypic variability of HOS, genetic testing will also be useful in guiding the clinical management of mildly affected individuals since even those without structural heart disease are at risk for cardiac conduction disease (3). Furthermore, families with an identified TBX5 mutation can be counseled about reproductive options, including preimplantation genetic diagnosis, for future pregnancies (30). For individuals not meeting clinical criteria for HOS, more appropriate genetic testing should be considered. Other disorders to be considered in individuals with limb and cardiac malformation include Okihiro syndrome, UMS, thrombocytopenia absent radius syndrome, and VACTERL association. Appropriate definition and diagnosis of the clinical features of heart-hand syndromes like HOS will allow us to better understand the role of their causative genes in development and ultimately will foster improved molecularly based therapeutic approaches for affected individuals.
Abbreviations
- ASD:
-
atrial septal defects
- CHD:
-
congenital heart disease
- dHPLC:
-
denaturing HPLC
- HOS:
-
Holt-Oram syndrome
- FHx:
-
family history
- TBX:
-
T-box
- VACTERL:
-
vertebral anomalies, anal atresia, cardiac malformations, tracheoesophageal fistula, renal anomalies, limb anomalies
- VSD:
-
ventricular septal defects
References
Elek C, Vitez M, Czeizel E 1991 [Holt-Oram syndrome]. Orv Hetil 132: 73–74, 77-78
Holt M, Oram S 1960 Familial heart disease with skeletal malformations. Br Heart J 22: 236–242
Basson CT, Cowley GS, Solomon SD, Weissman B, Poznanski AK, Traill TA, Seidman JG, Seidman CE 1994 The clinical and genetic spectrum of the Holt-Oram syndrome (heart-hand syndrome). N Engl J Med 330: 885–891
Newbury-Ecob RA, Leanage R, Raeburn JA, Young ID 1996 Holt-Oram syndrome: a clinical genetic study. J Med Genet 33: 300–307
Allanson JE, Newbury-Ecob RA 2003 Holt-Oram syndrome: is there a “face”?. Am J Med Genet A 118: 314–318
Basson CT, Bachinsky DR, Lin RC, Levi T, Elkins JA, Soults J, Grayzel D, Kroumpouzou E, Traill TA, Leblanc-Straceski J, Renault B, Kucherlapati R, Seidman JG, Seidman CE 1997 Mutations in human TBX5 cause limb and cardiac malformation in Holt-Oram syndrome. Nat Genet 15: 30–35
Li QY, Newbury-Ecob RA, Terrett JA, Wilson DI, Curtis AR, Yi CH, Gebuhr T, Bullen PJ, Robson SC, Strachan T, Bonnet D, Lyonnet S, Young ID, Raeburn JA, Buckler AJ, Law DJ, Brook JD 1997 Holt-Oram syndrome is caused by mutations in TBX5, a member of the Brachyury (T) gene family. Nat Genet 15: 21–29
Bamshad M, Le T, Watkins WS, Dixon ME, Kramer BE, Roeder AD, Carey JC, Root S, Schinzel A, Van Maldergem L, Gardner RJ, Lin RC, Seidman CE, Seidman JG, Wallerstein R, Moran E, Sutphen R, Campbell CE, Jorde LB 1999 The spectrum of mutations in TBX3: genotype/phenotype relationship in Ulnar-Mammary syndrome. Am J Hum Genet 64: 1550–1562
Bongers EM, Duijf PH, van Beersum SE, Schoots J, Van Kampen A, Burckhardt A, Hamel BC, Losan F, Hoefsloot LH, Yntema HG, Knoers NV, van Bokhoven H 2004 Mutations in the human TBX4 gene cause small patella syndrome. Am J Hum Genet 74: 1239–1248
Braybrook C, Doudney K, Marcano AC, Arnason A, Bjornsson A, Patton MA, Goodfellow PJ, Moore GE, Stanier P 2001 The T-box transcription factor gene TBX22 is mutated in X-linked cleft palate and ankyloglossia. Nat Genet 29: 179–183
Yagi H, Furutani Y, Hamada H, Sasaki T, Asakawa S, Minoshima S, Ichida F, Joo K, Kimura M, Imamura S, Kamatani N, Momma K, Takao A, Nakazawa M, Shimizu N, Matsuoka R 2003 Role of TBX1 in human del22q11.2 syndrome. Lancet 362: 1366–1373
Brassington AM, Sung SS, Toydemir RM, Le T, Roeder AD, Rutherford AE, Whitby FG, Jorde LB, Bamshad MJ 2003 Expressivity of Holt-Oram syndrome is not predicted by TBX5 genotype. Am J Hum Genet 73: 74–85
Cross SJ, Ching YH, Li QY, Armstrong-Buisseret L, Spranger S, Lyonnet S, Bonnet D, Penttinen M, Jonveaux P, Leheup B, Mortier G, Van Ravenswaaij C, Gardiner CA 2000 The mutation spectrum in Holt-Oram syndrome. J Med Genet 37: 785–787
Heinritz W, Moschik A, Kujat A, Spranger S, Heilbronner H, Demuth S, Bier A, Tihanyi M, Mundlos S, Gruenauer-Kloevekorn C, Froster UG 2005 Identification of new mutations in the TBX5 gene in patients with Holt-Oram syndrome. Heart 91: 383–384
Clough MV, Hamlington JD, McIntosh I 1999 Restricted distribution of loss-of-function mutations within the LMX1B genes of nail-patella syndrome patients. Hum Mutat 14: 459–465
Akrami SM, Winter RM, Brook JD, Armour JA 2001 Detection of a large TBX5 deletion in a family with Holt-Oram syndrome. J Med Genet 38: E44
Terrett JA, Newbury-Ecob R, Cross GS, Fenton I, Raeburn JA, Young ID, Brook JD 1994 Holt-Oram syndrome is a genetically heterogeneous disease with one locus mapping to human chromosome 12q. Nat Genet 6: 401–404
Bonnet D, Terrett J, Pequignot-Viegas E, Weissenbach J, Munnich A, Lyonnet S, Kachaner J 1995 Gene localization in 12q12 in Holt-Oram atrio-digital syndrome. Arch Mal Coeur Vaiss 88: 661–666
Fryns JP, Bonnet D, De Smet L 1996 Holt-Oram syndrome with associated postaxial and central polydactyly. Further evidence for genetic heterogeneity in the Holt-Oram syndrome. Genet Couns 7: 323–324
Vaughan CJ, Hom Y, Okin DA, McDermott DA, Lerman BB, Basson CT 2003 Molecular genetic analysis of PRKAG2 in sporadic Wolff-Parkinson-White syndrome. J Cardiovasc Electrophysiol 14: 263–268
Fan C, Duhagon MA, Oberti C, Chen S, Hiroi Y, Komuro I, Duhagon PI, Canessa R, Wang Q 2003 Novel TBX5 mutations and molecular mechanism for Holt-Oram syndrome. J Med Genet 40: e29
Basson CT, Huang T, Lin RC, Bachinsky DR, Weremowicz S, Vaglio A, Bruzzone R, Quadrelli R, Lerone M, Romeo G, Silengo M, Pereira A, Krieger J, Mesquita SF, Kamisago M, Morton CC, Pierpont ME, Muller CW, Seidman JG, Seidman CE 1999 Different TBX5 interactions in heart and limb defined by Holt-Oram syndrome mutations. Proc Natl Acad Sci U S A 96: 2919–2924
Basson CT 2000 Holt-Oram syndrome vs. heart-hand syndrome. Circulation 101: E191
Basson CT, Solomon SD, Weissman B, MacRae CA, Poznanski AK, Prieto F, Ruiz de la Fuente S, Pease WE, Levin SE, Holmes LB, Seidman JG, Seidman CE 1995 Genetic heterogeneity of heart-hand syndromes. Circulation 91: 1326–1329
Benson DW, Sharkey A, Fatkin D, Lang P, Basson CT, McDonough B, Strauss AW, Seidman JG, Seidman CE 1998 Reduced penetrance, variable expressivity, and genetic heterogeneity of familial atrial septal defects. Circulation 97: 2043–2048
Towbin JA, Bowles NE 2002 The failing heart. Nature 415: 227–233
Moss C, Bacon CJ, Mueller RF 1990 “Isolated” radial ray defect may be due to Rothmund-Thomson syndrome. Clin Genet 38: 318–319
Kohlhase J, Schubert L, Liebers M, Rauch A, Becker K, Mohammed SN, Newbury-Ecob R, Reardon W 2003 Mutations at the SALL4 locus on chromosome 20 result in a range of clinically overlapping phenotypes, including Okihiro syndrome, Holt-Oram syndrome, acro-renal-ocular syndrome, and patients previously reported to represent thalidomide embryopathy. J Med Genet 40: 473–478
Hatcher CJ, Goldstein MM, Mah CS, Delia CS, Basson CT 2000 Identification and localization of TBX5 transcription factor during human cardiac morphogenesis. Dev Dyn 219: 90–95
He J, McDermott DA, Song Y, Gilbert F, Kligman I, Basson CT 2004 Preimplantation genetic diagnosis of human congenital heart malformation and Holt-Oram syndrome. Am J Med Genet 126: 93–98
Acknowledgements
The authors thank the patients and their physicians for their contribution to this study. We also thank Dr. Peter Okin for his critical insights.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
McDermott, D., Bressan, M., He, J. et al. TBX5 Genetic Testing Validates Strict Clinical Criteria for Holt-Oram Syndrome. Pediatr Res 58, 981–986 (2005). https://doi.org/10.1203/01.PDR.0000182593.95441.64
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/01.PDR.0000182593.95441.64
This article is cited by
-
Genomic and physiological analyses of the zebrafish atrioventricular canal reveal molecular building blocks of the secondary pacemaker region
Cellular and Molecular Life Sciences (2021)
-
Functional analysis of two novel TBX5 variants present in individuals with Holt–Oram syndrome with different clinical manifestations
Molecular Genetics and Genomics (2021)
-
Holt-Oram syndrome: clinical and molecular description of 78 patients with TBX5 variants
European Journal of Human Genetics (2019)
-
Clinical and epidemiological features of Heart-Hand Syndrome: a hospital-based study in China
Scientific Reports (2018)
-
Malfunction defended
Synthese (2017)
