
Abstract
The placenta is essential to nutrition before birth. Recent work has shown that a range of clearly defined alterations can be found in the placentas of infants with intrauterine growth restriction (IUGR). In the mouse, a placental specific knockout of a single imprinted gene, encoding IGF-2, results in one pattern of alterations in placenta structure and function which leads to IUGR. We speculate that the alterations in the human placenta can also be grouped into patterns, or phenotypes, that are associated with specific patterns of fetal growth. Identifying the placental phenotypes of different fetal growth patterns will improve the ability of clinicians to recognize high-risk patients, of laboratory scientists to disentangle the complexities of IUGR, and of public health teams to target interventions aimed at ameliorating the long-term adverse effects of inadequate intrauterine growth.
Similar content being viewed by others
Main
The placenta is central to human development and in particular to fetal nutrition. However, research on this organ has been limited because, with some exceptions (1), the placenta is generally regarded as a passive sieve, or conduit, that does not provide clinically relevant information about the condition of the fetus. This view cannot be sustained in the light of recent research that shows that the placenta can give specific indications about the degree of fetal compromise; that placental nutrient transfer is altered if the fetus is smaller than expected, i.e. in cases of IUGR, and that the placenta can actively regulate the nature and extent of nutrient transfer to the fetus.
These observations are particularly important because of the increasing recognition that growth in early life has implications for health in later life. For example, the incidence of diabetes, hypertension, and obesity (metabolic syndrome) and the risk of ischemic heart disease are strongly related to growth before birth (2,3). Furthermore, IUGR (which may be defined as the failure of a fetus to reach its genetic growth potential) is associated with poor outcome in the neonatal period, especially among infants born prematurely (4,5). Reducing the adverse effects of poor antenatal growth will be a central component of attempts to improve the health status of future generations. These efforts will benefit from a comprehensive understanding of how the placenta behaves in IUGR.
In this review, we briefly describe alterations in morphology, blood flow, and fetal oxygen delivery found in placentas from human pregnancies complicated by IUGR. We proceed by reviewing new data suggesting that human IUGR is a condition associated with specific alterations in placental nutrient transporter expression and activity. Exciting recent work in mice provides an elegant illustration of how placental gene expression, morphology, and nutrient transport can contribute to delineating a distinct phenotype. Mouse studies also emphasize the likely importance of imprinted genes in this regard. Some alterations found in human placentas in pregnancies affected by IUGR are analogous to those found in mice and are highly likely to also represent elements of clinically relevant phenotypes. Robust placental phenotyping would advance the ability of the clinician to separate a pathologically IUGR baby from one that is healthy but genetically small or to distinguish between IUGR and other antenatal adversities such as in utero infection. Furthermore, we note that research into the associations between poor growth and ill health has generally focused on organ systems damaged at the end of the disease pathway, including the heart and brain. Since the pathway to poor growth and ill health appears to start with the placenta in many cases, placental phenotypes of IUGR will provide an opportunity to focus research efforts on the causes of growth restriction rather than the effects.
PLACENTAL MORPHOLOGY
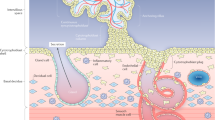
In the human placenta, the primary barrier to maternal-fetal exchange is the syncytiotrophoblast, a transporting epithelium covering the placental villi which project into the maternal blood of the intervillous space (Fig. 1). Villi contain capillary networks derived from the fetal circulation. Recent studies of placental morphology have demonstrated distinct abnormalities of villi that could be associated with particular presentations of IUGR (6,7). For example, one abnormal pattern of villous morphology (straight villi with a simple vascular network and fewer interconnections than normal) is found in severe, early-onset preeclampsia and is associated with altered results on Doppler imaging of flow through the umbilical arteries (discussed below) (8). A contrasting pattern of placental villous morphology (villi are more tortuous and have more interconnections than normal) is not as frequently associated with abnormal umbilical hemodynamics or severe preeclampsia and is thought to reflect alterations in placental development late in gestation (8). A more general morphologic observation in IUGR, important because of likely effects on diffusional permeability of the placenta, is that the surface area of the syncytiotrophoblast is reduced whereas the thickness of the exchange barrier formed by the trophoblast and fetal capillary endothelium is increased (9). Additionally, IUGR of all types shows evidence of increased placental apoptosis (10), reflecting altered cell turnover in the placenta, which could be associated with the changes in the size and internal architecture of the organ.

Morphology of the villi and exchange barrier in the normal human placenta. (a) Cross section of the placenta in situ: maternal blood is supplied by the spiral arteries and bathes the villi in the intervillous space. (b) A villous tree. The villi are the functional units of the placenta, covered in an epithelium called the syncytiotrophoblast; the umbilical artery, running in the cord, supplies fetal blood to the fetoplacental capillary network investing the core of each villous and draining into the umbilical vein. (c) An electron micrograph of the exchange barrier consisting of syncytiotrophoblast, the matrix of the villous core, and the fetal capillary endothelium. The surface area of the exchange barrier is proportional to the number of villous buds and the degree of expansion provided by the microvilli on the maternal facing plasma membrane of the syncytiotrophoblast. Diffusional transfer will be indirectly proportional to the thickness of the barrier (double-headed arrow: corresponds to 3 μm). Transfer capacity will also be affected by the number and activity of transporter proteins (•) in the microvillous and basal plasma membranes. (Micrograph courtesy of Dr. C.J.P. Jones).
PLACENTAL BLOOD FLOW, OXYGEN, AND NUTRIENT DELIVERY
Clinical studies of IUGR have included a long-standing focus on blood flow through the maternal and fetal circulations of the placenta. Doppler velocimetry analysis has shown reduced blood flow in both the uterine and umbilical arteries in IUGR, and such measurements, particularly in regard to the umbilical circulation, have made a significant contribution to recent advances in the management of high-risk pregnancies (11). In normal pregnancies, branches of the uterine arteries are converted into low-resistance uteroplacental vessels. Alterations in this process have been observed on placental bed biopsies from IUGR pregnancies and are associated with evidence of bilateral high-resistance flow velocity waveforms with early diastolic notches at 24 weeks of gestation (11,12). However, abnormal patterns of uterine artery Doppler signal show poor sensitivity and specificity for adverse outcome (13). Abnormal umbilical artery Doppler waveforms are thought to reflect deranged placental impedance secondary to altered vessel morphology in the villi (see above). Absent or reversed end-diastolic flow velocity in the umbilical artery waveforms shows a strong association with increased perinatal mortality (11).
In utero cord blood sampling has demonstrated that IUGR is associated with hypoxemia (14). Under normal circumstances, oxygen diffuses readily across barriers and exchange is primarily limited by the rate of supply to, and removal from, the barrier, i.e. blood flow (15). However, in IUGR uterine venous samples show significantly higher O2 content and lower coefficients of uterine O2 extraction (16). This suggests, like the Doppler studies, that impairment of the fetoplacental circulation in the villi and consequent decrease in the ability of the fetus to extract oxygen are of considerably greater importance etiologically than changes in uterine blood flow. Furthermore, in all cases of IUGR, fetuses have lower plasma concentrations of amino acids than infants of normal size (17,18). The physicochemical characteristics of these nutrients mean that their rate of transfer is much slower than that of oxygen and is limited much more by the exchange barrier itself than by blood flow (15). Together, these data and the morphologic observations described above focus attention in IUGR on the development of the capacity of the villi, and specifically the syncytiotrophoblast, to supply nutrients to the fetus.
PLACENTAL TRANSPORT
An abundance of descriptive data, primarily obtained in vitro, has recently accumulated describing changes in placental transport capacity in pregnancies complicated by IUGR due to alterations in activity of plasma membrane transporter proteins. In humans and animal models, IUGR is typified by a decrease in the activity of placental amino acid transporters. The System A transporter (expressed by genes SLC38-A1, -A2 and, -A4) is critical in mediating the uptake of neutral amino acids (glycine/serine/alanine) across the syncytiotrophoblast maternal facing, microvillous plasma membrane (MVM) (Fig. 1), the rate-limiting step in transplacental amino acid transfer (19). Within the normal range of birth weights, an inverse relationship between birth weight and MVM System A activity has been demonstrated (20). By contrast, MVM System A activity has consistently been shown to be down-regulated in IUGR (21–24). Furthermore, the most severe cases of IUGR, as assessed by abnormal pulsatility index in the umbilical artery and abnormal fetal heart rate tracings, are associated with the most pronounced decreases in MVM System A activity (23). These data suggest that reduced MVM System A activity is a robust marker for IUGR. The transport of essential amino acids, such as taurine (25), leucine, and lysine (26) has also been reported to be decreased in the MVM and/or fetal facing, basal plasma membrane (BM) isolated from IUGR placentas. These in vitro findings are compatible with data on cord plasma amino acid concentrations (17,18) and an in vivo study in pregnant women, using stable isotopes, which showed that placental transfer of the essential amino acids leucine and phenylalanine is reduced in IUGR (27).
Transport of other solutes is also affected. The activity of MVM lipoprotein lipase is critical in releasing free fatty acids (FFA) incorporated in lipoproteins circulating in maternal blood. Thus, lipoprotein lipase–mediated breakdown of lipoproteins is the important first step in transplacental transfer of FFA. MVM lipoprotein lipase activity is reduced in IUGR (28), consistent with clinical studies showing lower fetal/maternal ratios for long-chain polyunsaturated fatty acids in IUGR (29). Placental ion transport is either correlated with fetal growth or appears to be regulated in a compensatory manner relative to fetal growth. Both Na+/K+ ATPase and Na+/H+ exchanger (NHE) placental expression and activity are down-regulated in human IUGR (23,30–32). These two critical membrane transport systems are involved in pH regulation, vectorial Na+ transport and maintenance of the Na+ gradient that drives the transport of other vital nutrients such as amino acids. Alterations in MVM NHE activity appear to be related to the severity of IUGR: transporter activity is not changed in less severe IUGR cases, primarily at term (22,30,32). Some ions appear to be regulated quite differently. BM Ca2+ ATPase (or calcium pump) is up-regulated in cases of IUGR, which may represent a compensatory activation of the placental calcium transport system stimulated by fetal demand (33).
Placental transporters can be regulated by a variety of factors that are likely to be clinically relevant. Hypoxia reduces the expression and activity of System A amino acid transporter (34) and increases the expression of glucose transporters in cultured term human trophoblasts (35). The placenta itself may participate in the regulation of transporters in an autocrine/paracrine fashion. For example, leptin is produced in large amounts by the syncytiotrophoblast (36), leptin receptors are present in the syncytiotrophoblast plasma membranes, and leptin stimulates System A activity in isolated villous fragments in vitro (37). In addition, small-for-gestational age babies have low umbilical plasma leptin concentrations at birth (38). Furthermore, insulin and the insulinlike growth factors may affect placental nutrient transfer by increasing the activity of the System A transporter (37,39). In human IUGR, umbilical plasma concentrations of PTH-related peptide (PTHrp) 38–94 are elevated (33) and PTHrp 38–94 stimulates BM Ca2+ ATPase activity in vitro (40), providing one possible mechanism underlying the increased activity of the calcium pump in human IUGR (33).
It is important to note that some transporters do not show altered activity in IUGR. For example, both the expression of the glucose transporter GLUT1, per unit membrane area, and the glucose transporter activity in MVM and BM, are unchanged in IUGR (24,41). This indicates that the type of alterations in transporter activity seen for System A are not found for all transporters and are not a general reaction to adversity. We can summarize these data by classifying alterations to placental transporters in IUGR to three groups: (1) nutrient transporters that are unaffected by IUGR, e.g. glucose; (2) transporter activity that is decreased when fetal growth is restricted e.g. System A, NHE; (3) changes in transport activity potentially compensatory to reduced fetal and placental growth e.g. Ca2+.
This classification reflects the data in relation to human IUGR as reviewed above. There may be differences in other species. For example, expression of a second glucose transporter isoform, GLUT3, as well as GLUT1 may be important in the sheep placenta (42). GLUT3 but not GLUT1 expression is reduced in placentas from globally undernourished rat mothers, in which the fetuses are growth restricted (43), whereas in the same species both GLUT1 and GLUT3 are up-regulated in dexamethasone-induced IUGR (44).
CAUSE AND EFFECT: HELP FROM THE MOUSE
Implicit in much of the above data is a difficulty inherent in understanding the relationship between placental phenotypes and IUGR: does the former cause or follow from the latter? There are hints in the data described already that the placental phenotypes in IUGR are a mixture of both scenarios. However, recent work on a knockout mouse model in which the placental-specific transcript of the IGF-2 (Igf-2) gene was deleted shows unequivocally that changes that create a placental phenotype, similar to a group of those found in human IUGR, can precede a decline in fetal growth (45,46). In this mutant mouse, placental weight is reduced as early as d 14 of pregnancy (term is d 20 in this species, so this equates to about 28/40 weeks in human), but fetal growth restriction is not found until about d 19 (45). The reasons for this delay in restriction to fetal growth seem to be twofold (45,46): (1) initial up-regulation then decline in System A activity in the mutant placentas and (2) an increasingly marked reduction in the diffusional permeability of the mutant placentas to hydrophilic molecules, apparently arising from a decrease in placental trophoblast surface area and an increase in exchange barrier thickness that is remarkably similar to that found in the human placenta in IUGR (9).
The Igf-2 gene is imprinted, expressed only from the paternal allele. Approximately 60 imprinted genes have so far been discovered, and all that have been examined appear to have a role in controlling placental and fetal growth (47). Interestingly these imprinted genes include hormonal regulators, such as Igf-2, and transporters themselves: the System A SLC38–A4 gene has recently been shown to be expressed only from the paternal allele (48,49). This suggests that examination of expression of imprinted genes could be an important component of placental phenotyping.
CONCLUSIONS
These observations, accumulated essentially over the past decade, have enabled us to describe a series of very specific alterations to an organ that many perceive as homogeneous in health and disease. This constellation of findings is particularly striking given the multifactorial etiology of IUGR, difficulties in distinguishing genetically small from growth restricted fetuses, and differing experimental methodologies. Furthermore, the information from the placenta-specific igf-2 knockout mouse model of IUGR has shown how a combination of these alterations can be related to altered expression of a single gene. Thus, in mouse, a multifaceted placental phenotype resulting in IUGR is related to a single gene deletion. This leads us to speculate that defining similar phenotypes in humans (not necessarily related to the IGF system) will help to disentangle the complexities of IUGR. We propose that a rigorously defined placental phenotype be taken as the starting point for defining IUGR and other fetal growth patterns rather than vice versa. This is represented diagrammatically in Figure 2. As shown in Figure 2A, the fetus who is of normal size for gestational age (AGA) and of normal size for its genetic potential (AGP) is appropriately grown and has a placenta with a normal phenotype (phenotype 1). An alternative explanation for an AGA infant is that the infant is small for its genetic potential (SGP). Hitherto, clinical examination has not allowed these two possibilities to be distinguished reliably but in the case illustrated (Fig. 2A), the AGA:SGP infant has a moderately abnormal phenotype (e.g. decreased exchange surface area, increased thickness, reduced expression/activity of amino acid transporters [phenotype 2]). In Figure 2B, a situation is depicted in which a small for gestational age infant may be AGP or SGP. The SGA:SGP infant is likely to be identifiable by an abnormal placental phenotype (phenotype 4 compared with phenotype 3). Finally, as shown in Figure 2C, the IUGR baby delivered prematurely due to severe fetal compromise has a placental phenotype that is markedly abnormal (phenotype 6) and different from the placental phenotypes associated with other causes of prematurity. As well as the general risks of prematurity (phenotype 5), a premature infant with IUGR will have specific risks derived from in utero malnutrition.

Size at birth and short- and long-term risks. Concept diagram showing how knowledge of placental phenotype could lead to improved classification of the “small” baby and the “normal” sized baby. Three idealized possible (54) clinical presentations are represented, based on the size of the infant: normal (a), small (b), and very small (c), together with idealized in utero growth patterns and outcomes for six distinct patterns that could be revealed by placental phenotyping (phenotypes 1–6). See text for further description.
We suggest that infants with abnormal placental phenotypes have a greater risk of neonatal mortality and morbidity and possibly ischemic heart failure and diabetes in later life. Currently, the distinctions between infants who are AGP/SGP are difficult, and we propose that clinical markers derived from detailed placental phenotyping may aid the clinician in the identification of babies at risk of short- and long-term consequences. For example, in the mouse, a reduction in placental size was an early indicator of IUGR (45,46) and in women, the three-dimensional ultrasound measurement of placental volume in the second trimester correlates significantly with birth weight (50,51). Placental volume measurements may therefore provide one predictor of later decreases in fetal growth rate. The predictive value of such clinically applicable measures is likely to be improved when they are integrated with novel tests for other placental phenotypes. Development of such novel tests is the exciting prospect that can now be glimpsed. Possibilities include measurement of transporter activity in the fragments of MVM plasma membranes, which are shed into maternal blood during every pregnancy (52) or the use of positron emission tomography with suitable tracer substrates to image placental transporter activity in utero: a suitable substrate for System A ([N-methyl-11C]alpha-methylaminoisobutyric acid) is now available (53). These specific diagnostic tests are essential prerequisites for developing the currently nonexistent in utero therapies for fetal growth anomalies, which are the ultimate goal. Placental phenotyping both in utero and after delivery will provide more predictive information concerning diseases in later life than the crude proxies of intrauterine growth currently available.
Abbreviations
- AGA:
-
appropriate for gestational age
- AGP:
-
appropriate for genetic potential
- BM:
-
basal plasma membrane
- IGF-2:
-
insulin-like growth factor-2
- IUGR:
-
intrauterine growth restriction
- MVM:
-
microvillous plasma membrane
- NHE:
-
Na+/H+ exchanger
- SGP:
-
small for genetic potential
References
Hay WW Jr 1991 Not just a conduit for maternal fuels. Diabetes 40( suppl 2): 44–50
Barker DJ 1992 Fetal and Infant Origins of Adult Disease. BMJ Press, London, pp 315–322
Hack M, Flannery DJ, Schluchter M, Cartar L, Borawski E, Klein N 2002 Outcomes in young adulthood for very-low-birth-weight infants. N Engl J Med 346: 149–157
Bernstein IM, Horbar JD, Badger GJ, Ohlsson A, Golan A 2000 Morbidity and mortality among very-low-birth-weight neonates with intrauterine growth restriction. The Vermont Oxford Network. Am J Obstet Gynecol 182: 198–206
Resnik R 2002 Intrauterine growth restriction. Obstet Gynecol 99: 490–496
Kingdom J, Huppertz B, Seaward G, Kaufmann P 2000 Development of the placental villous tree and its consequences for fetal growth. Eur J Obstet Gynecol Reprod Biol 92: 35–43
Mayhew TM, Charnock-Jones DS, Kaufmann P 2004 Aspects of human fetoplacental vasculogenesis and angiogenesis. III. Changes in complicated pregnancies. Placenta 25: 127–139
Todros T, Sciarrone A, Piccoli E, Guiot C, Kaufmann P, Kingdom J 1999 Umbilical Doppler waveforms and placental villous angiogenesis in pregnancies complicated by fetal growth restriction. Obstet Gynecol 93: 499–503
Mayhew TM, Ohadike C, Baker PN, Crocker IP, Mitchell C, Ong SS 2003 Stereological investigation of placental morphology in pregnancies complicated by pre-eclampsia with and without intrauterine growth restriction. Placenta 24: 219–226
Smith SC, Baker PN, Symonds EM 1997 Increased placental apoptosis in intrauterine growth restriction. Am J Obstet Gynecol 177: 1395–1401
Karsdorp VH, van Vugt JM, van Geijn HP, Kostense PJ, Arduini D, Montenegro N, Todros T 1994 Clinical significance of absent or reversed end diastolic velocity waveforms in umbilical artery. Lancet 344: 1664–1668
Pijnenborg R, Bland JM, Robertson WB, Brosens I 1983 Uteroplacental arterial changes related to interstitial trophoblast migration in early human pregnancy. Placenta 4: 397–414
Aardema MW, Oosterhof H, Timmer A, van Rooy I, Aarnoudse JG 2001 Uterine artery Doppler flow and uteroplacental vascular pathology in normal pregnancies and pregnancies complicated by pre-eclampsia and small for gestational age fetuses. Placenta 22: 405–411
Pardi G, Cetin I, Marconi AM, Lanfranchi A, Bozzetti P, Ferrazzi E, Buscaglia M, Battaglia FC 1993 Diagnostic value of blood sampling in fetuses with growth retardation. N Engl J Med 328: 692–696
Schroder HJ 1995 Comparative aspects of placental exchange functions. Eur J Obstet Gynecol Reprod Biol 63: 81–90
Pardi G, Cetin I, Marconi AM, Bozzetti P, Buscaglia M, Makowski EL, Battaglia FC 1992 The venous drainage of the human uterus: respiratory gas studies in normal and fetal growth retarded pregnancies. Am J Obstet Gynecol 166: 699–706
Cetin I, Marconi AM, Bozzetti P, Sereni LP, Corbetta C, Pardi G, Battaglia FC 1988 Umbilical amino acid concentrations in appropriate and small-for-gestational age infants: a biochemical difference present in utero. Am J Obstet Gynecol 158: 120–126
Cetin I, Ronzoni S, Marconi AM, Perugino G, Corbetta C, Battaglia FC, Pardi G 1996 Maternal concentrations and fetal-maternal concentration differences of plasma amino acids in normal and intrauterine growth-restricted pregnancies. Am J Obstet Gynecol 174: 1575–1583
Jansson T 2001 Amino acid transporters in the human placenta. Pediatr Res 49: 141–147
Godfrey KM, Matthews N, Glazier J, Jackson A, Wilman C, Sibley CP 1998 Neutral amino acid uptake by microvillous plasma membrane of the human placenta is inversely related to fetal size at birth in normal pregnancy. J Clin Endocrinol Metab 83: 3320–3326
Dicke JM, Henderson GI 1988 Placental amino acid uptake in normal and complicated pregnancies. Am J Med Sci 295: 223–227
Mahendran D, Donnai P, Glazier JD, D'Souza SW, Boyd RD, Sibley CP 1993 Amino acid (System A) transporter activity in microvillous membrane vesicles from the placentas of appropriate and small for gestational age babies. Pediatr Res 34: 661–665
Glazier JD, Cetin I, Perugino G, Ronzoni S, Grey AM, Mahendran D, Marconi AM, Pardi G, Sibley CP 1997 Association between the activity of the system A amino acid transporter in the microvillous plasma membrane of the human placenta and severity of fetal compromise in intrauterine growth restriction. Pediatr Res 42: 514–519
Jansson T, Ylven K, Wennergren M, Powell TL 2002 Glucose transport and system A activity in syncytiotrophoblast microvillous and basal membranes in intrauterine growth restriction. Placenta 23: 386–391
Norberg S, Powell TL, Jansson T 1998 Intrauterine growth restriction is associated with a reduced activity of placental taurine transporters. Pediatr Res 44: 233–238
Jansson T, Scholtbach V, Powell TL 1998 Placental transport of leucine and lysine is reduced in intrauterine growth restriction. Pediatr Res 44: 532–537
Paolini CL, Marconi AM, Ronzoni S, Di Noio M, Fennessey PV, Pardi G, Battaglia FC 2001 Placental transport of leucine, phenylalanine, glycine, and proline in intrauterine growth-restricted pregnancies. J Clin Endocrinol Metab 86: 5427–5432
Magnusson AL, Waterman IJ, Wennergren M, Jansson T, Powell TL 2004 Triglyceride hydrolase activities and expression of fatty acid binding proteins in human placenta in pregnancies complicated by intrauterine growth restriction and diabetes. J Clin Endocrinol Metab 89: 4607–4614
Cetin I, Giovannini N, Alvino G, Agostoni C, Riva E, Giovannini M, Pardi G 2002 Intrauterine growth restriction is associated with changes in polyunsaturated fatty acid fetal-maternal relationships. Pediatr Res 52: 750–755
Johansson M, Jansson T, Glazier JD, Powell TL 2002 Activity and expression of the Na+/H+ exchanger is reduced in syncytiotrophoblast microvillous plasma membranes isolated from preterm intrauterine growth restriction pregnancies. J Clin Endocrinol Metab 87: 5686–5694
Johansson M, Karlsson L, Wennergren M, Jansson T, Powell TL 2003 Activity and protein expression of Na+K+ ATPase are reduced in microvillous syncytiotrophoblast plasma membranes isolated from pregnancies complicated by intrauterine growth restriction. J Clin Endocrinol Metab 88: 2831–2837
Hughes JL, Doughty IM, Glazier JD, Powell TL, Jansson T, D'Souza SW, Sibley CP 2000 Activity and expression of the Na+/H+ exchanger in the microvillous plasma membrane of the syncytiotrophoblast in relation to gestation and small for gestational age birth. Pediatr Res 48: 652–659
Strid H, Bucht E, Jansson T, Wennergren M, Powell T 2003 ATP-dependent Ca2+ transport across basal membrane of human syncytiotrophoblast in pregnancies complicated by diabetes or intrauterine growth restriction. Placenta 24: 445–452
Nelson DM, Smith SD, Furesz TC, Sadovsky Y, Ganapathy V, Parvin CA, Smith CH 2003 Hypoxia reduces expression and function of system A amino acid transporters in cultured term human trophoblasts. Am J Physiol Cell Physiol 284: C310–C315
Esterman A, Greco MA, Mitani Y, Finlay TH, Ismail-Beigi F, Dancis J 1997 The effect of hypoxia on human trophoblast in culture: morphology, glucose transport and metabolism. Placenta 18: 129–136
Linnemann K, Malek A, Sager R, Blum WF, Schneider H, Fusch C 2000 Leptin production and release in the dually in vitro perfused human placenta. J Clin Endocrinol Metab 85: 4298–4301
Jansson N, Greenwood S, Johansson BR, Powell TL, Jansson T 2003 Leptin stimulates system A activity in human placental villous fragments. J Clin Endocrinol Metab 88: 1205–1211
Harigaya A, Nagashima K, Nako Y, Morikawa A 1997 Relationship between concentration of serum leptin and fetal growth. J Clin Endocrinol Metab 82: 3281–3284. Erratum in: J Clin Endocrinol Metab 84:3106
Bloxam DL, Bax BE, Bax CM 1994 Epidermal growth factor and insulin-like growth factor I differently influence the directional accumulation and transfer of 2-aminoisobutyrate (AIB) by human placental trophoblast in two-sided culture. Biochem Biophys Res Commun 199: 922–929
Strid H, Care A, Jansson T, Powell T 2002 Parathyroid hormone-related peptide (38–94) amide stimulates ATP-dependent calcium transport in the basal plasma membrane of the human syncytiotrophoblast. J Endocrinol 175: 517–524
Jansson T, Wennergren M, Illsley NP 1993 Glucose transporter protein expression in human placenta throughout gestation and in intrauterine growth retardation. J Clin Endocrinol Metab 77: 1554–1562
Das UG, He J, Ehrhardt RA, Hay WW Jr, Devaskar SU 2000 Time-dependent physiological regulation of ovine placental GLUT-3 glucose transporter protein. Am J Physiol Regul Integr Comp Physiol 279: R2252–R2261
Lesage J, Hahn D, Leonhardt M, Blondeau B, Breant B, Dupouy JP 2002 Maternal undernutrition during late gestation-induced intrauterine growth restriction in the rat is associated with impaired placental GLUT3 expression, but does not correlate with endogenous corticosterone levels. J Endocrinol 174: 37–43
Langdown ML, Sugden MC 2001 Enhanced placental GLUT1 and GLUT3 expression in dexamethasone-induced fetal growth retardation. Mol Cell Endocrinol 185: 109–117
Constancia M, Hemberger M, Hughes J, Dean W, Ferguson-Smith A, Fundele R, Stewart F, Kelsey G, Fowden A, Sibley C, Reik W 2002 Placental-specific IGF-II is a major modulator of placental and fetal growth. Nature 417: 945–948
Sibley CP, Coan PM, Ferguson-Smith AC, Dean W, Hughes J, Smith P, Reik W, Burton GJ, Fowden AL, Constancia M 2004 Placental-specific insulin-like growth factor 2 (Igf2) regulates the diffusional exchange characteristics of the mouse placenta. Proc Natl Acad Sci U S A 101: 8204–8208
Reik W, Constancia M, Fowden A, Anderson N, Dean W, Ferguson-Smith A, Tycko B, Sibley C 2003 Regulation of supply and demand for maternal nutrients in mammals by imprinted genes. J Physiol 547: 35–44
Mizuno Y, Sotomaru Y, Katsuzawa Y, Kono T, Meguro M, Oshimura M, Kawai J, Tomaru Y, Kiyosawa H, Nikaido I, Amanuma H, Hayashizaki Y, Okazaki Y 2002 Asb4, Ata3, and Dcn are novel imprinted genes identified by high-throughput screening using RIKEN cDNA microarray. Biochem Biophys Res Commun 290: 1499–1505
Smith RJ, Dean W, Konfortova G, Kelsey G 2003 Identification of novel imprinted genes in a genome-wide screen for maternal methylation. Genome Res 13: 558–569
Hafner E, Philipp T, Schuchter K, Dillinger-Paller B, Philipp K, Bauer P 1998 Second trimester measurement of placental volume by 3D ultrasound to predict SGA-infants. Ultrasound Obstet Gynecol 12: 97–102
Kinare AS, Natekar AS, Chinchwadkar MC, Yajnik CS, Coyaji KJ, Fall CH, Howe DT 2000 Low midpregnancy placental volume in rural Indian women: A cause for low birth weight. Am J Obstet Gynecol 182: 443–448
Knight M, Redman CW, Linton EA, Sargeant IL 1998 Shedding of syncytiotrophoblast microvilli into the maternal circulation in preeclamptic pregnancies. Br J Obstet Gynaecol 105: 632–640
Sutinen E, Jyrkkio S, Alanen K, Nagren K, Minn H 2003 Uptake of [N-methyl-11C]alpha-methylaminoisobutyric acid in untreated head and neck cancer studied by PET. Eur J Nucl Med Mol Imaging 30: 72–77
Sparks JW, Ross JC, Cetin I 1998 Intrauterine growth and nutrition. In: Polin RA, Fox WW (eds) Fetal and Neonatal Physiology. WB Saunders, Philadelphia, pp 267–289
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Sibley, C., Turner, M., Cetin, I. et al. Placental Phenotypes of Intrauterine Growth. Pediatr Res 58, 827–832 (2005). https://doi.org/10.1203/01.PDR.0000181381.82856.23
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/01.PDR.0000181381.82856.23
This article is cited by
-
Sexually dimorphic patterns in maternal circulating microRNAs in pregnancies complicated by fetal growth restriction
Biology of Sex Differences (2021)
-
Hypoxia and oxidative stress induce sterile placental inflammation in vitro
Scientific Reports (2021)
-
Vascular changes in fetal growth restriction: clinical relevance and future therapeutics
Journal of Perinatology (2019)
-
NLRP7 is increased in human idiopathic fetal growth restriction and plays a critical role in trophoblast differentiation
Journal of Molecular Medicine (2019)
-
Identification of placental nutrient transporters associated with intrauterine growth restriction and pre-eclampsia
BMC Genomics (2018)
