
Abstract
The mitochondrial trifunctional protein (TFP) is a multienzyme complex of the β-oxidation cycle. Human TFP is an octamer composed of four α-subunits harboring long-chain enoyl-CoA hydratase and long-chain L-3-hydroxyacyl-CoA dehydrogenase and four β-subunits encoding long-chain 3-ketoacyl-CoA thiolase. Mutations in either subunit may result in general TFP deficiency with reduced activity of all three enzymes. We report five new patients with α-subunit mutations and compare general TFP deficiency caused by α-subunit mutations (n = 15) to that caused by β-subunit mutations (n = 13) with regard to clinical features, enzyme activity, mutations, thiolase expression, and thiolase protein turnover. Among patients with α-subunit mutations, the same three heterogeneous phenotypes reported in patients with β-subunit mutations were observed: a lethal form with predominating cardiomyopathy; an infancy-onset, hepatic presentation; and a milder, later-onset, neuromyopathic form. Maternal HELLP syndrome (hemolysis, elevated liver enzymes, low platelets) occurred with an incidence of 15 to 20%, as in families with β-subunit mutations. Enzyme assays in fibroblasts revealed an identical biochemical pattern in both groups. α-Subunit mutational analysis demonstrated molecular heterogeneity, with 53% (9 of 17) truncating mutations. In contrast, patients with β-subunit mutations had predominantly missense mutations. Thiolase expression in fibroblasts was as markedly reduced in α-subunit patients as in the β-subunit group with similarly increased thiolase degradation, presumably secondary to TFP complex instability. TFP deficiency as a result of either α- or β-subunit mutations presents with similar, heterogeneous phenotypes. Both α- and β-subunit mutations result in TFP complex instability, demonstrating that the mechanism of disease is the same in α- or β-mutation-derived disease and explaining the biochemical and clinical similarities.
Similar content being viewed by others


Main
Mitochondrial trifunctional protein (TFP) deficiency is a rare genetic disorder of the fatty acid β-oxidation (FAO) cycle. The human trifunctional protein is an octamer composed of four α-subunits and four β-subunits (1, 2). The α-subunit contains the 2,3-long-chain enoyl CoA hydratase and the long-chain 3-hydroxy-acyl CoA dehydrogenase (LCHAD) domains; the β-subunit harbors the long-chain 3-keto acyl CoA thiolase (LKAT) domain. These enzymes catalyze the last three steps of the β-oxidation spiral of long-chain fatty acids. General TFP deficiency is defined by reduced activity of all three TFP enzymes. Mutations resulting in TFP deficiency lie in either the α- or the β-subunit, but the mechanisms by which mutations in one subunit reduce activities of all three enzymes have not been studied extensively. The TFP subunits are encoded by separate nuclear genes located in the same region of chromosome 2p23 (3).
The most common defect of the TFP complex is isolated LCHAD deficiency (LCHADD), which is defined biochemically by reduced LCHAD activity with substantial preservation of the other two TFP enzymatic activities. E474Q in the α-subunit is the common mutation for this disorder and is located within the catalytic region of the LCHAD domain (4). It is present on 60%(5) to 86%(6) of abnormal alleles in LCHADD, and more than 60 patients with isolated LCHADD and this mutation have been reported (5–7). Despite the occurrence of a common mutation, LCHADD presents with heterogeneous phenotypes (6).
General TFP deficiency has been reported much less frequently, most often with infantile mortality secondary to severe cardiomyopathy. However, more recently, TFP deficiency as a result of β-subunit mutations has been characterized as a phenotypically and molecularly heterogeneous disorder, with a predominance of mild manifestations (8). Three different types of presentations have been apparent in general TFP deficiency with β-subunit mutations, termed the lethal phenotype, the hepatic phenotype, and the neuromyopathic phenotype (8). The lethal phenotype is characterized by severe dilated cardiomyopathy, lactic acidosis, a Reye syndrome-like picture of hypoketotic hypoglycemia, and neonatal death. The hepatic phenotype is defined as a less severe presentation with onset in the first months of life characterized by episodes of hypoketotic hypoglycemia and lethargy usually triggered by illness or fasting. The main features of the neuromyopathic form with later onset are progressive peripheral neuropathy and episodic, often exercise-induced myoglobinuria (8).
General TFP deficiency as a result of α-subunit mutations has been reported in 10 families (1, 5, 9–15). Six presented with the lethal phenotype (1, 5, 12–15); neuromyopathic forms have been reported in four (9–11). Because of the clinical similarities among patients with TFP deficiency caused by either α- or β-subunit mutations, we postulated that similar pathogenic mechanisms exist in both groups. We have collected five more patients with general TFP deficiency and α-subunit mutations and characterize the overall total group of 15 individuals with α-mutation-derived disease, including the 10 already reported (1, 5, 9–15), with regard to clinical features, mutations, enzyme activities, thiolase expression in fibroblasts, and TFP complex stability.
METHODS
We report five new patients with TFP deficiency and α-subunit mutations. Patients' clinical and family histories were obtained from the referring physicians. Because all patients' TFP deficiency was diagnosed and evaluated in different metabolic centers, there were no common diagnostic and treatment protocols. Diagnosis was confirmed by enzyme and/or molecular analysis.
To investigate the mechanism of disease in patients with α- and β-subunit mutations, we analyzed TFP subunit expression by immunoblot of patient-derived fibroblast protein extracts and quantified LKAT stability using pulse-chase studies in these cells. All studies were performed with the approval of the Vanderbilt University Medical Center or Washington University Institutional Review Boards. The studies were conducted with informed consent from the parents.
Cell lines.
Fibroblast cell lines were maintained in Dulbecco's modified Eagle's medium (GIBCO Invitrogen Corporation, www.invitrogen.com) with 10% fetal bovine serum, 20 mM of glutamine, antibiotics, and nonessential amino acids.
Enzyme analysis.
General TFP deficiency is defined by reduced activities of all three enzymes to demarcate it from isolated LCHADD. LKAT is encoded on the β-subunit and LCHAD on the α-subunit, and deficiency of both activities is always associated with a lack of activity of 2,3-long-chain enoyl CoA hydratase (1, 16).
Fibroblasts for LCHAD and LKAT enzyme assays were available in four of the five patients' newly diagnosed TFP deficiency. Enzyme activities were measured according to previously reported techniques (7). Healthy individuals served as control subjects.
PCR amplification and sequencing analysis.
DNA was isolated from whole blood or fibroblasts by standard techniques (17). All 20 α-subunit and all 16 β-subunit exons of patients' genomic DNA were amplified in the presence of (32P)-dCTP by PCR under standard conditions (15). Details about the amplification of α-subunit exons, including oligonucleotide sequences of the primer pairs, have previously been reported (15). For the amplification of β-subunit exons, we designed 23- to 27-bp-long intronic primer pairs, based on our own and previously reported genomic thiolase DNA sequences (18). We directly sequenced exonic DNA of all α- and β-subunit exons using the reported intronic primer pairs.
Immunoblot analysis.
Western blot analysis was performed after 4 to 20% SDS-PAGE (19) using rabbit polyclonal antibodies raised against recombinant β-subunit TFP (thiolase) or mitochondrial malate dehydrogenase (mMDH), a tricarboxylic acid cycle enzyme. Because mMDH expression is constant, the ratio between thiolase and mMDH antigen allows comparison of protein loading. Extracts from fibroblasts cultured from a healthy individual served as the control.
Pulse-chase analysis and immunoprecipitation.
Fibroblasts were labeled with 2 mL of 0.1 mCi/mL [35S] methionine per 1 × 107 cells for 1 h as previously described (20, 21). After this pulse, the medium was changed to fresh Dulbecco's modified Eagle's medium with antibiotics and fetal bovine serum and incubated for 2, 4, 8, 24, and 48 h, respectively, the “chase” period as previously described (1). Thiolase and mMDH were immunoprecipitated by standard techniques (22). As an internal control, extracts from the healthy individual's fibroblasts were treated with preimmune serum to determine nonspecifically precipitated proteins. The labeled products were subject to SDS-PAGE and fluorography to quantify radiolabeled β-subunit and mMDH expression.
With the computer program Scion image (www.scioncorp.com), the area and the density of scanned pulse-chase images were quantified to determine the amount of thiolase protein antigen present at the different time points. The data were plotted against the time to determine thiolase decay in all available fibroblast lines from TFP-deficient patients, one LCHAD-deficient patient, and one healthy individual. Protein half-life was calculated from the exponential trend line that was laid through the measured time points. The half-life was t1/2 = ln(0.5):k.
RESULTS
Phenotypes of TFP-deficient patients with α-subunit mutations.
Among the five patients with α-subunit mutations not previously reported, two died in the neonatal period with severe cardiomyopathy, one presented with the hepatic phenotype, and one presented with the neuromyopathic phenotype. The mother of patient 5 developed acute fatty liver of pregnancy (AFLP) in the 36th week that led to the diagnosis in the newborn. This patient was born with an arterial cord pH of 7.06 and was artificially ventilated immediately after birth. He recovered quickly and over 4 y has not developed any clinical TFP-associated symptoms while being treated with preventive measures, including avoidance of fasting and a reduced long-chain fat diet. Autopsy in one patient with the lethal form revealed severe lipid accumulation in many organs, including the myocardium, the renal tubules, and the liver. Severe cardiomyopathy with pericardial and pleural effusions was observed in this patient. The hepatic presentation in one patient was characterized by episodes of hypoketotic hypoglycemia with onset at 4.5 mo of age, accompanied by seizures. The patient with the myopathic presentation developed severe muscular hypotonia and respiratory failure at 9 mo of age precipitated by an infection. Shortness of breath, intermittent oxygen dependence, and hypoventilation during metabolic decompensation have been observed before in patients with TFP deficiency and β-subunit mutations, suggesting weakness of the diaphragm (8).
Six of the 10 previously reported patients with α-subunit mutations presented with the lethal, cardiac phenotype and died in the neonatal period (1, 5, 12–15). The other four became symptomatic with later-onset myopathy (9–11). Three had proven peripheral neuropathy. In one individual peripheral neuropathy was not observed up to the age of 12 y (10); however, retinitis pigmentosa was apparent in this patient at age 3.
Maternal liver diseases such as HELLP (hemolysis, elevated liver enzymes, low platelets) syndrome and AFLP were observed in three of these overall 15 families (20%). Onset of symptoms in this cumulative total of 15 patients with α-subunit mutations ranged from the first day of life to 11 years of age. The age at diagnosis was between the first week of life and 13 y. In comparison, among the 13 reported TFP-deficient patients with β-subunit mutations, disease onset was between the first day of life and 6 y of age, and the age at diagnosis ranged from the first week of life to 18 y of age (8).
Thus, the clinical manifestations of general TFP deficiency as a result of α-subunit mutations are very similar to TFP deficiency with β-subunit mutations (8). However, the distribution of phenotypes differs in both groups. A total of 53% of patients with α-subunit mutations presented with the lethal phenotype, whereas 70% of patients with β-subunit mutations had milder phenotypes (8).
LCHAD and LKAT activities in fibroblasts.
LCHAD activities in our four α-subunit patients (patients 4, 6, 9, and 11;Fig. 1) ranged from 21 to 34% of normal (mean, 27.6%; SD, ±4.9%; median, 27.9%), LKAT activities were 6 to 36% of normal (mean, 22.9%; SD, ±12.3%; median, 24.9%). Residual activities are relatively high in some patients and might be due to other enzymes with overlapping substrate specificity in our in vitro assay system. Different levels of activity of these other enzymes may account for the variable residual activities between patients in our in vitro assays.

Patients 1 to 5 present with the lethal phenotype, patient 6 with the hepatic form, and patients 7 through 10 with the neuromyopathic phenotype. Patient 11 remained asymptomatic during follow-up until 4 y of age. Patients 4, 6, 9, and 11 are reported for the first time. LCHAD and LKAT activities were determined in fibroblast cells. Patients were collected over a number of years, and assays in patients and batch normals were performed at different times. Each patient was compared with five batch normals that were examined at the same time. Activities are given in percentage of the normal. Patient 1 did not present with any measurable LKAT activity. Patients 8, 9, and 11 carry the E474Q mutation on one allele.
In 11 of the total of 15 reported patients with TFP deficiency and α-subunit mutations, enzyme results are available (Fig. 1). A comparison between residual activities in patients with α- and β-subunit mutations is shown in Figure 2, demonstrating that both groups are biochemically similar.

LCHAD and LKAT activities in patients with α- or β-subunit mutations are given as median and are subdivided according to phenotype subgroups. These data demonstrate similar biochemical effects as a result of α- or β-subunit mutations. Mild TFP deficiency includes patients with the hepatic and the neuromyopathic phenotypes.
Patient 1 of the α-subunit group (Fig. 1) presented with an exceptionally high LCHAD activity of 50%. However, no thiolase activity was measurable in this patient (Fig. 3), clearly demonstrating the effect of the homozygous α-subunit mutations (IVS6 −2A>G) on β-subunit expression.

Immunoblot analysis of thiolase in comparison with mMDH is demonstrated in six different cell lines as further described in the text. The subunit that carries the mutations is indicated. The patients with milder TFP deficiency have at least one missense mutation. β-Subunit missense mutations at the R-28 residue are considered severe because of its location in the dimerization domain of the thiolase protein (8). The mutation on the other allele was not delineated in this patient.
Molecular genetic analysis.
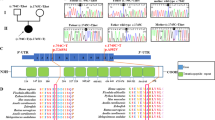
The diagnosis was confirmed by α-subunit molecular genetic analysis in all five new patients. Four different missense mutations (E474Q, V563M, D665G, and V376L), one premature termination mutation (Y-13ter), and two different splice site mutations (IVS18 +1G>A and IVS4 −2A>T) were delineated (Table 1). Two patients were homozygous for the mutations found, and the other three were compound heterozygotes. The E474Q mutation was present in two unrelated patients on one allele.
Among the cumulative total of 15 patients with α-subunit mutations and general TFP deficiency, four presented with the E474Q mutation on one allele (9, 10, our patients). The mutations on the other allele were IVS18 +1G>A, R524ter, and D665G. In one patient (8), the second mutation was not found and might be in intronic regions not contained within our amplified PCR products.
The mutational spectrum of all reported patients with α- and β-subunit mutations is summarized in Table 1. The molecular results show that null mutations (9 of 17) are more common in the α-subunit group, and this correlates with the more common occurrence of lethal and early-onset hepatic phenotypes among these patients. In contrast, missense mutations predominate in the β-subunit group (12 of 16), consistent with the higher proportion of individuals with the mild, myopathic phenotype in this group. The molecular data demonstrate both that heterogeneous α- and β-subunit mutations result in general TFP deficiency and that missense mutations in either subunit can result in biochemical deficiency of all three α- and β-subunit enzymes.
Thiolase protein expression in fibroblasts.
To investigate the pathogenetic mechanism by which mutations in either TFP subunit cause general TFP deficiency, we investigated LKAT, the enzyme encoded by the β-subunit, both by immunoblot to quantify steady-state expression of protein and by pulse-chase analyses to quantify the rate of subunit degradation. Because both subunits are required for stable expression of the TFP octamer (1), these experiments are a measure of TFP complex stability.
Thiolase expression was assessed by Western blot using protein extracts from fibroblast cell lines derived from patients with lethal and mild TFP deficiency as a result of α-or β-subunit mutations (Fig. 3). Patients with mild TFP deficiency represent individuals with the neuromyopathic phenotype. All patients with general TFP deficiency presented with reduced or undetectable cellular thiolase levels in comparison with the control cell line. The patient with isolated LCHADD had normal cellular thiolase protein, whereas in both TFP-deficient patients exhibiting the lethal phenotype, thiolase protein was either absent or barely detectable; reduction in thiolase protein in cells from patients with the mild phenotype was also severe but less than in lethal phenotypes. Thus, correlation of level of thiolase antigen expression with phenotype was apparent in both groups of TFP deficiency. These data demonstrate that mutations in either TFP subunit affect β-subunit thiolase expression to a similar extent.
Thiolase stability and degradation.
Thiolase degradation rates were determined in the same fibroblast cell lines using radiolabeling of proteins and pulse-chase methods. mMDH again served as an internal control. On fluorography, the thiolase and mMDH bands are clearly identified because their molecular weights are 35 and 51 kD, respectively (Fig. 4). The third band that was apparent on SDS-PAGE is nonspecific, because it is also present in the sample incubated only with preimmune serum (Fig. 4). The amount of thiolase protein was plotted against the time to determine thiolase decay (Fig. 5). According to our calculations from the exponential trend lines through the data points, thiolase half-life was 43 h in a healthy individual and 55 h in an LCHAD-deficient patient. In patients with TFP deficiency, thiolase half-life was significantly reduced to 8.8 h in mild α-subunit mutated TFP deficiency and 5.6 h in mild β-subunit mutated TFP deficiency. The severe forms presented with half-lives of 1.3 h (α-subunit mutated) and 2.2 h (β-subunit mutated), respectively (Fig. 5). Thus, thiolase degradation is much more rapid in all TFP-deficient patients in comparison with a healthy individual or the LCHAD-deficient patient. Mutations in the α- or β-subunit can affect β-subunit breakdown to similar extents. The degree of change in thiolase degradation rates correlates nicely with the severity of phenotype, with very rapid loss in cells from patients with lethal, early-onset disease and intermediate rates of degradation in milder cases with the neuropathic phenotype.

After a 1-h pulse with [35S] methionine, cell lines from each patient were incubated with nonlabeled medium for various times. With immunoprecipitation, thiolase and mMDH were selected and are identified on fluorography by their molecular weights. Thiolase (▸) represents the upper band; mMDH represents the lower band. The intermediate band is nonspecific, because it is also present in a control incubated with preimmune serum. With the computer program Scion image, the signal intensity of each band was calculated to determine degradation rates (Fig. 5).

The amount of thiolase antigen detected on fluorography (Fig. 4) was plotted against the time to determine thiolase decay in all available fibroblast lines from TFP-deficient patients, one LCHAD-deficient patient with the homozygous E474Q mutation, and a healthy individual. Protein half-life was calculated from the exponential trend line that was laid through the measured time points. Thiolase stability is affected in all TFP-deficient patients in comparison with the control, and the degradation rates correlate with the phenotype in both groups.
DISCUSSION
This study demonstrates that mutations in the TFP α-subunit reduce expression and increase breakdown of the β-subunit protein, the LKAT, that is part of the octameric TFP. β-Subunit mutations have similar effects on expression and degradation. That mutations in either subunit equally alter TFP complex stability is consistent with our observations that TFP deficiency as a result of α- or β-subunit mutations is clinically and biochemically similar.
In general TFP deficiency as a result of α-subunit mutations, three forms of disease manifestation have become apparent, just as we described in TFP deficiency as a result of β-subunit mutations (8), and phenotype profiles in both groups are similar in regard to symptoms and disease onset. All patients with α- or β-subunit mutations and the early-onset phenotype died in the neonatal period despite intensive care treatment. This observation contrasts to very long-chain acyl-CoA dehydrogenase (VLCAD) deficiency, another long-chain FAO defect, that also presents with three phenotypes of differing severities (23). However, the early-onset, cardiac phenotype in VLCAD deficiency is not always lethal. In fact, life-threatening cardiomyopathy and pericardial effusion in VLCAD deficiency are completely reversible within weeks after implementation of treatment measures (24–26).
Fifty-three percent of patients with TFP deficiency and α-subunit mutations presented with this lethal phenotype. However, 57% of the total of 28 known patients with general TFP deficiency survived long-term, and their disease was well controlled after implementation of preventive measures such as avoidance of fasting and a reduced long-chain fat intake. Therefore, the widespread impression that general TFP deficiency is a fatal disease is incorrect. A previous survey revealed that among 107 patients with FAO defects, 53% survived, suggesting a similar distribution of mild and severe phenotypes (27). General TFP deficiency as a result of α-subunit mutations is therefore clinically as heterogeneous as TFP deficiency secondary to β-subunit mutations and as other FAO defects.
Two clinical features are unique to disorders that affect the TFP complex. First, the occurrence of peripheral neuropathy has not been observed in other FAO defects but is common among survivors with general TFP and isolated LCHAD deficiencies (6). Retinitis pigmentosa has previously been reported only in isolated LCHADD (27, 28) but is a long-term complication in TFP deficiency as well, as one of the patients from our study demonstrated (10). Second, maternal liver diseases, such as HELLP syndrome and AFLP, are complications in 15 to 20% of pregnancies with TFP-deficient fetuses, whether the mutation site is in either the α- or β-subunit. Maternal liver disease is relatively common with an incidence of 19%(6) to 79%(5, 29) in isolated LCHADD families with the common E474Q mutation, but HELLP and AFLP are very rare in pregnancies of fetuses with other FAO defects, such as medium-chain acyl-CoA dehydrogenase deficiency.
The distribution of phenotypes in patients with α- or β-subunit mutations is reflected well by the molecular data, with a predominance of null mutations in α-subunit patients and a predominance of missense mutations in β-subunit patients (8) (Table 1). Both groups are characterized by molecular heterogeneity, and only a few mutations are shared among unrelated patients (6 of 33 mutations). In four unrelated patients of the α-subunit group, the E474Q mutation that usually results in isolated LCHADD was present on one allele. In three of these patients, fibroblasts for enzyme studies were available and revealed clearly reduced LKAT activities of 17 to 33% of normal. LCHAD activities ranged from 21 to 28% (Table 1). Clinically, three of these four patients presented with a neuromyopathic phenotype, and one patient received the diagnosis as a result of maternal AFLP and was asymptomatic at the age of 4 y. We speculate that in TFP-deficient patients with a heterozygous E474Q mutation, the second mutation has a significant dominant-negative effect on protein structure and stability because patients homozygous for the E474Q mutation exhibit a normal thiolase half-life. Because this common LCHAD mutation may occur in the heterozygous state in patients with general TFP deficiency, our current practice of defining TFP and LCHAD deficiencies enzymatically allows a more correct disease classification than molecular data alone. Enzyme assays in lymphocytes quickly provide results and may be the suitable first-line investigation in cases of acylcarnitine profiles suggestive of TFP deficiency.
By enzyme data, patient 1 (Fig. 1) might be more correctly defined as having isolated thiolase deficiency that has not been previously reported. It is interesting that this patient is homozygous for a splice site mutation in the α-subunit (IVS6 −2A>G), and the lack of the α-subunit likely causes loss of the β-subunit. Thiolase expression was severely reduced on immunoblot in this patient as a result of an increased thiolase degradation rate (Figs. 3 to 5, example for severe α-TFP). The high residual LCHAD activities in this patient may be due to other enzymes with overlapping substrate specificity, especially short-chain 3-hydoxy-acyl-CoA dehydrogenase in our assays.
All α- and β-subunit mutations found in patients with general TFP deficiency, even though 60% are missense mutations, alter TFP complex sufficiently to severely reduce activities of both α- and β-subunit enzymes. Patients with α-subunit mutations associated with rapidly degraded β-subunit protein in fibroblasts demonstrate the significant effect of the α-subunit mutations on the whole octameric TFP complex. Therefore, either α- or β-subunit mutations can cause accelerated thiolase degradation rates. In contrast, isolated LCHAD deficiency as a result of homozygous E474Q mutations is associated with normal thiolase stability and expression. The mutation site is within the LCHAD catalytic domain and does not have a major effect on LCHAD structure.
CONCLUSION
In conclusion, TFP α- and β-subunit mutations have comparable effects on TFP complex expression and thiolase degradation. Thus, intact α- and β-subunits are equally essential for stabilizing the TFP complex (1, 2). Thiolase expression levels and the different rates of thiolase degradation as a result of either α- or β-subunit mutations correlate well with the severity of clinical manifestations. Because the mechanism of disease is the same in TFP deficiency as a result of α- or β-subunit mutations, the biochemical and clinical phenotype is similar in both groups.
Abbreviations
- TFP:
-
trifunctional protein
- FAO:
-
fatty acid oxidation
- LCHAD:
-
long-chain 3-hydroxy acyl-CoA dehydrogenase
- LKAT:
-
long-chain 3-ketoacyl-CoA thiolase
- LCHADD:
-
LCHAD deficiency
- mMDH:
-
mitochondrial malate dehydrogenase
- AFLP:
-
acute fatty liver of pregnancy
- HELLP:
-
hemolysis, elevated liver enzymes, low platelets
- VLCAD:
-
very long-chain acyl-CoA dehydrogenase
References
Ushikubo S, Aoyama T, Kamijo T, Wanders RJ, Rinaldo P, Vockley J, Hashimoto T 1996 Molecular characterization of mitochondrial trifunctional protein deficiency: formation of the enzyme complex is important for stabilization of both α- and β-subunits. Am J Hum Genet 58: 979–988
Weinberger MJ, Rinaldo P, Strauss AW, Bennett MJ 1995 Intact α-subunit is required for membrane-binding of human mitochondrial trifunctional β-oxidation protein, but is not necessary for conferring 3-ketoacyl-CoA thiolase activity to the β-subunit. Biochem Biophys Res Commun 209: 47–52
Yang BZ, Heng HH, Ding JH, Roe CR 1996 The genes for the α- and β-subunits of the mitochondrial trifunctional protein are both located in the same region on human chromosome 2p23. Genomics 37: 141–143
Barycki JJ, O'Brien LK, Bratt JM, Zhang R, Sanishvili R, Strauss AW, Banaszak LJ 1999 Biochemical characterization and crystal structure determination of human heart short chain L-3-hydroxyacyl-CoA dehydrogenase provide insights into catalytic mechanism. Biochem 38: 5786–5798
Ibdah JA, Bennett MJ, Rinaldo P, Zhao Y, Gibson B, Sims HF, Strauss AW 1999 A fetal fatty-acid oxidation disorder as a cause of liver disease in pregnant women. N Engl J Med 340: 1723–1731
Den Boer ME, Wanders RJ, Morris AA, Ijlst L, Heymans HS, Wijburg FA 2002 Long-chain 3-hydoxyacyl-CoA dehydrogenase deficiency: clinical presentation and follow-up of 50 patients. Pediatrics 109: 99–104
Wanders RJ, Vreken P, den Boer ME, Wijburg FA, van Gennip AH, Ijlst L 1999 Disorders of mitochondrial fatty acyl-CoA β-oxidation. J Inherit Metab Dis 22: 442–487
Spiekerkoetter U, Sun B, Khuchua Z, Bennett M, Strauss AW 2003 Molecular and phenotypic heterogeneity in mitochondrial trifunctional protein deficiency due to β-subunit mutations. Hum Mutat 21: 598–607
Schaefer J, Jackson S, Dick DJ, Turnbull DM 1996 Trifunctional enzyme deficiency: adult presentation of a usually fatal β-oxidation defect. Ann Neurol 40: 597–602
Isaacs JD, Sims HF, Powell CK, Bennett MJ, Hale DE, Treem WR, Strauss AW 1996 Maternal acute fatty liver of pregnancy associated with fetal trifunctional protein deficiency: molecular characterization of a novel maternal mutant allele. Pediatr Res 40: 393–398
Ibdah JA, Tein I, Dionisi-Vici C, Bennett MJ, Ijist LI, Gibson B, Wanders RJ, Strauss AW 1998 Mild trifunctional protein deficiency is associated with progressive neuropathy and myopathy and suggests a novel genotype-phenotype correlation. J Clin Invest 102: 1193–1199
Matern D, Strauss AW, Hillman SL, Mayatepek E, Millington DS, Trefz FK 1999 Diagnosis of mitochondrial trifunctional protein deficiency in a blood spot from the newborn screening card by tandem mass spectrometry and DNA analysis. Pediatr Res 46: 45–49
Hintz SR, Matern D, Strauss AW, Bennett MJ, Hoyme E, Schelley S, Kobori J, Colby C, Lehman NL, Enns GM 2002 Early neonatal diagnosis of long-chain 3-hydroxy acyl coenzyme A dehydrogenase and mitochondrial trifunctional protein deficiencies. Mol Genet Metab 75: 120–127
Spiekerkoetter U, Eeds A, Yue Z, Haines J, Strauss AW, Summar M 2002 Uniparental disomy of chromosome 2 resulting in lethal trifunctional protein deficiency due to homozygous α-subunit mutations. Hum Mutat 20: 447–451
Brackett JC, Sims HF, Rinaldo P, Shapiro S, Powell CK, Bennett MJ, Strauss AW 1995 Two α-subunit donor splice site mutations cause human trifunctional protein deficiency. J Clin Invest 95: 2076–2082
Jackson S, Kler RS, Bartlett K, Pourfarzam M, Aynsley-Green A, Bindoff LA, Turnbull DM 1992 Combined enzyme defect of mitochondrial fatty acid oxidation. J Clin Invest 90: 1219–1225
Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K 1994 Current Protocols in Molecular Biology. John Wiley & Sons, New York
Kamijo T, Aoyama T, Komiyama A, Hashimoto T 1994 Structural analysis of cDNA for subunits of human mitochondrial fatty acid β-oxidation trifunctional protein. Biochem Biophys Res Commun 199: 818–825
Laemmli UK 1970 Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227: 680–685
Kamijo T, Wanders RJA, Saudubray JM, Aoyama T, Komiyama A, Hashimoto T 1994 Mitochondrial trifunctional protein deficiency: catalytic heterogeneity of the mutant enzyme in two patients. J Clin Invest 93: 1740–1747
Pulse-chase labeling of cells with [35S] methionine 2002 Current protocols in Cell Biology Online. John Wiley and Sons. Available at: www.mrw2.interscience.wiley.com
Immunoprecipitation using cells in suspension lysed with a nondenaturing detergent solution 2002 Current protocols in Cell Biology Online. John Wiley and Sons. Available at: www.mrw2.interscience.wiley.com
Gregersen N, Andresen BS, Corydon MJ, Corydon TJ, Olsen R, Bolund L, Bross P 2001 Mutation analysis in mitochondrial fatty acid oxidation defects exemplified by acyl-CoA dehydrogenase deficiencies, with special focus on genotype-phenotype relationship. Hum Mutat 18: 169–189
Brown-Harrison MC, Nada MA, Sprecher H, Vianey-Saban C, Farquhar J, Gilladoga AC, Roe CR 1996 Very long chain acyl-CoA dehydrogenase deficiency: successful treatment of cardiomyopathy. Biochem Mol Med 58: 59–65
Cox GF, Souri M, Aoyama T, Rockenmacher S, Varvogli L, Rohr F, Hashimoto T, Korson MS 1998 Reversal of severe hypertrophic cardiomyopathy and an excellent neurophysiologic outcome in very-long-chain acyl-coenzyme A dehydrogenase deficiency. J Pediatr 133: 247–253
Spiekerkoetter U, Tenenbaum T, Heusch A, Wendel U 2002 Cardiomyopathy and pericardial effusion in infancy point to a fatty acid β-oxidation defect after exclusion of an underlying infection. Pediatr Cardiol Dec 4 [epub ahead of print]
Saudubray JM, Martin D, de Lonlay P, Touati G, Poggi-Travert F, Bonnet D, Jouvet P, Boutron M, Slama A, Vianey-Saban C, Bonnefont JP, Rabier D, Kamoun P, Brivet M 1999 Recognition and management of fatty acid oxidation defects: a series of 107 patients. J Inherit Metab Dis 22: 488–502
Tyni T, Kivela T, Lappi M, Summanen P, Nikoskelainen E, Pihko H 1998 Ophthalmologic findings in long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency caused by the G1528C mutation: a new type of hereditary metabolic chorioretinopathy. Ophthalmology 105: 810–824
Yang Z, Zhao Y, Bennett MJ, Strauss AW, Ibdah JA 2002 Fetal genotypes and pregnancy outcomes in 35 families with mitochondrial trifunctional protein mutations. Am J Obstet Gynecol 187: 715–720
Author information
Authors and Affiliations
Corresponding author
Additional information
The study was supported by NIH grant P50 HL61006.
Rights and permissions
About this article
Cite this article
Spiekerkoetter, U., Khuchua, Z., Yue, Z. et al. General Mitochondrial Trifunctional Protein (TFP) Deficiency as a Result of Either α- or β-Subunit Mutations Exhibits Similar Phenotypes Because Mutations in Either Subunit Alter TFP Complex Expression and Subunit Turnover. Pediatr Res 55, 190–196 (2004). https://doi.org/10.1203/01.PDR.0000103931.80055.06
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/01.PDR.0000103931.80055.06
This article is cited by
-
A G1528C Hadha knock-in mouse model recapitulates aspects of human clinical phenotypes for long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency
Communications Biology (2023)
-
Fatal pitfalls in newborn screening for mitochondrial trifunctional protein (MTP)/long-chain 3-Hydroxyacyl-CoA dehydrogenase (LCHAD) deficiency
Orphanet Journal of Rare Diseases (2018)
-
Disorders of mitochondrial long-chain fatty acid oxidation and the carnitine shuttle
Reviews in Endocrine and Metabolic Disorders (2018)
-
Clinical and molecular investigation of 14 Japanese patients with complete TFP deficiency: a comparison with Caucasian cases
Journal of Human Genetics (2017)
-
Mitochondrial Fatty Acid Oxidation Disorders Associated with Cardiac Disease
Current Pathobiology Reports (2017)
