
Abstract
The PAX6 gene is a paradigm for our understanding of the molecular genetics of mammalian eye development. Twelve years after its identification it is one of the most intensively studied genes, both in terms of its diverse and complex functions during oculogenesis and its role in an ever-increasing variety of human congenital eye malformations. The PAX6 field has benefited greatly from the continued input of clinicians, human geneticists and developmental biologists. This review summarizes the latest data on the PAX6 mutation spectrum and recent insights into Pax6 function from the mouse.
Similar content being viewed by others


Main
PAX6 was identified as a candidate aniridia gene during the search for the genes responsible for the WAGR syndrome (Wilms tumor, aniridia, genitourinary malformations, and mental retardation), which is caused by hemizygous deletions of 11p13 (1). Heterozygous intragenic mutations were subsequently described in many nonsyndromic aniridia cases, confirming PAX6 as the aniridia gene (2–5). At the same time Pax6 mutations were found in the classical mouse microphthalmia mutant small eye(6).
The PAX6 protein is a transcriptional regulator with two highly conserved DNA binding domains, a paired domain and a homeodomain (1, 3, 7, 8;Fig. 1). The homeodomain is followed by a proline, serine, and threonine-rich trans-activation domain (9). The last 30 amino acids constitute a highly conserved C-terminal peptide that modulates DNA binding by the homeodomain (10).

The PAX6 cDNA and protein. The cDNA is represented as a horizontal bar (top). Vertical lines indicate exon boundaries (3). PAX6 has 10 constitutive coding exons, 4–13, and one alternatively spliced exon, 5a. Inclusion of exon 5a results in the insertion of 14 amino acids into the paired domain. PB, paired box (red); LNK, linker region; HB, homeobox (yellow); PST, proline, serine, threonine-rich domain; CT, C-terminal region (green). A graphical representation of the PAX6 protein (bottom) shows the different domains of the translated protein. The paired domain (red) and the homeodomain (yellow) are shown in contact with DNA. The homeodomain consists of 3 α-helices (parallelograms linked by ribbons) while the paired domain consists of 6 α-helices. The structures of the PST domain and the C-terminal peptide are unknown. N and C indicate the N- and C-termini of the protein, respectively.
THE SPECTRUM OF HUMAN PAX6 MUTATIONS
Molecular analyses of WAGR syndrome cases showed that aniridia can be caused by deletion of one copy of PAX6(1, 11). This led to the proposal that aniridia results from PAX6 haploinsufficiency and is caused by loss-of-function of one copy of the gene (1). Once a significant number of intragenic mutations had been reported (2–5) it was clear that the classical aniridia phenotype could be caused by mutations throughout the PAX6 coding region. The simplest explanation for the observation that deletions and intragenic mutations cause the same phenotype is that both types of mutation are functionally null (4, 5, 12).
Human PAX6 mutations are archived in the PAX6 Allelic Variant Database, http://pax6.hgu.mrc.ac.uk/ (13). The database contains 292 records of which 275 refer to pathologic mutations in the coding region of the gene. Fig. 2 shows a breakdown of these 275 mutations by class.

The PAX6 mutation spectrum. Pie-chart showing the different mutation types among 275 pathologic mutations in the PAX6 coding region. N, nonsense mutation; FS, frame-shifting insertion or deletion; S, splice mutation; M, missense mutation; AT, anti-termination mutation; IF, in-frame insertion or deletion.
The most common PAX6 mutations are the nonsense mutations R240X (21 reports), R317X (15 reports), and R203X (15 reports). These mutations, which involve CpG dinucleotides, account for 50% of all nonsense mutations and 19% of all PAX6 mutations.
Frameshift, nonsense, and splice mutations, which together make up 71% of PAX6 mutations, would all be predicted to introduce a premature termination codon into the PAX6 reading frame. The resulting mutant mRNAs are likely to be detected by RNA surveillance and degraded by nonsense-mediated decay (14–16). RNA surveillance is a powerful mechanism to prevent the accumulation of truncated peptides that could interfere with the function of the normal protein or have toxic effects in the cell.
“Anti-termination” mutations – where the stop codon is changed to a coding codon – have recently emerged as a significant new mutation class (10, 17–20). The consequence of such a mutation is predicted to be continuation of translation into the 3′ untranslated region of the PAX6 gene. The C-terminus of the PAX6 protein, including the position of the stop codon, is highly conserved between species and functional studies show that the C-terminal region is important for DNA binding by the homeodomain (10). Addition of a peptide encoded by the 3′ untranslated region would be expected to disrupt the function of the C-terminus. Of 11 anti-termination mutations in the database, 8 are associated with aniridia and 3 are associated with milder iris defects (10, 19, 20). From these phenotypes it seems likely that anti-termination mutations generate alleles with partial or complete loss of function.
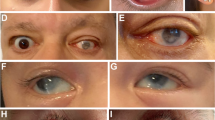
Missense mutations account for 18% of PAX6 mutations (Fig. 2). Missense mutations would not be detected by RNA surveillance and are predicted to result in a full-length PAX6 protein with a single amino acid substitution. Half of all missense mutations are associated with the aniridia phenotype and are therefore likely to have significant loss-of-function (20–23;Fig. 3). However missense mutations can potentially cause partial loss-of-function or gain-of-function. This may explain why the remaining 50% of missense mutations are associated with congenital eye malformations that are distinct from classical aniridia. These include isolated foveal hypoplasia, ectopia pupillae, and Peters' anomaly, a rare form of anterior segment dysgenesis characterized by central corneal opacification (23–28;Fig. 3). Two-thirds of missense mutations are in the paired domain and may affect the ability of the resultant protein to bind some, but not other, PAX6 targets. Indeed, differences in DNA binding activity and trans-activation activity have been reported for a number of paired domain missense mutant proteins (20, 21, 29).

Distinct ocular phenotypes associated with PAX6 missense mutations. (a) Aniridia with iris remnants (arrowheads) and congenital cataract (arrow) caused by the missense mutation A33P (23). (b) Ectopia pupillae caused by the missense mutation V126D (23). (c) Gonioscopic view of Peters anomaly, showing corneal opacification, caused by the missense mutation R26G (24). Panels (a) and (b) reproduced from Hanson et al.; 1999 Human Molecular Genetics vol 8 pp162–175, by permission of Oxford University Press. Panel (c) reproduced from Holmstrom et al. (1991) British Journal of Ophthalmology vol 75 pp591–597, by permission of the BMJ Publishing Group.
The spectrum of phenotypes associated with PAX6 missense mutations was extended recently with the report of missense mutations, including 5 in the PST domain, in a cohort of patients with a variety of optic nerve malformations including morning glory disc anomaly and optic nerve hypoplasia (29). The mutant proteins are impaired in their ability to auto-activate PAX6 and repress PAX2, which may affect the formation of the boundary between the optic cup and the optic stalk (see below).
Surprisingly the most highly conserved region of the PAX6 protein, the homeodomain, has just two reported missense mutations, one in an individual with a phenotype of very mild partial aniridia (30) and one in a patient with iris defects, coloboma of the optic nerve, retina and choroid, and persistent hyperplastic primary vitreous (29). The fact that the PAX6 homeodomain is very highly conserved throughout evolution argues that most changes in the amino acid sequence are selected against, presumably because they impair the function of the protein and cause a deleterious phenotype. Perhaps homeodomain missense mutations predominantly cause nonocular phenotypes, thus explaining the low frequency of reported mutations to date.
The PAX6 gene has one alternatively spliced exon, 5a (Fig. 1). Inclusion of exon 5a results in the insertion of 14 amino acids into the paired domain and changes the DNA binding specificity of the protein (31). Mutations that affect exon 5a cause congenital eye malformations in humans and mice (27, 31, 32).
ANIRIDIA AS PART OF THE WAGR SYNDROME
It is well known that congenital sporadic aniridia is associated with an elevated risk of later developing Wilms tumor (33). The molecular basis of this association is hemizygous deletions that remove one copy of PAX6– thus causing aniridia – and one copy of WT1– thus making it much more likely that a “second hit” in the remaining functional copy of WT1 will cause tumorigenesis (1, 11, 33). The PAX6 and WT1 genes lie 650 kb apart in 11p13 (http://www.ensembl.org/).
The proportion of sporadic aniridia cases with a deletion of both PAX6 and WT1 has been determined in a number of studies (18, 34, 35). Of 51 sporadic aniridia cases, 19 (37%) had a deletion of both genes. Of these 19 patients, 9 developed Wilms tumor. Thus the proportion of all sporadic cases developing Wilms tumor was 9/51 or 18%, while the proportion of deletion cases developing Wilms tumor was 9/19 = 47%. Wilms tumor was not observed in any nondeletion patients.
Familial aniridia patients may also carry deletions of the PAX6 gene (11) although the frequency is lower than in sporadic patients. The deletions are usually small and tend not to encompass the WT1 gene; however in one rare case a deletion of PAX6 and WT1 was passed from mother to son (36).
PAX6 deletions have been described in association with other eye anomalies. A child with bilateral Peters' anomaly had a deletion of WT1 and PAX6(24) and a child with congenital bilateral microphthalmia and severe anterior segment dysgenesis who later developed Wilms tumor had a deletion of 11p13–15.1 (37). A child with sporadic Rieger syndrome, including typical dental and maxillary anomalies, had a PAX6 deletion (38). Since PAX6 is not expressed in the developing teeth or maxilla, this individual may also have a mutation of PITX2, the Rieger syndrome gene, at 14q25 (39).
GENETIC TESTING AND COUNSELING
Most familial aniridia patients have mutations within the PAX6 gene, of the kind shown in Fig. 2. Aniridia is dominantly inherited with high penetrance. Affected individuals have a 50% chance of passing the mutant allele, and therefore the disease, to each offspring.
As mentioned above, about one-third of sporadic aniridia cases have a deletion of the WT1 and PAX6 genes and half of these will develop Wilms tumor (18, 34, 35). This emphasizes the importance of performing chromosomal deletion analysis in a newborn with sporadic aniridia. If WT1 is deleted, there is a significant risk of Wilms tumor and monitoring should be performed; however if no deletion is found the risk of Wilms tumor is reduced to that of the general population (33).
The remaining two-third of sporadic cases are most likely to have de novo mutations within the PAX6 gene, of the sort shown in Fig. 2. These mutations would be expected to be transmitted to the next generation in an autosomal dominant fashion.
EXTRA-OCULAR PHENOTYPES
Extra-ocular sites of PAX6 expression include the developing olfactory system, brain, neural tube, and endocrine pancreas (28, 40). A number of recent studies have highlighted nonocular phenotypes in aniridia patients.
Impaired olfaction is a form of sensory deprivation that is relatively overlooked in humans, but a recent study found that of 14 aniridia patients, all but one had impaired sense of smell, ranging from mild hyposmia to anosmia (19).
MRI scans of the brains of aniridia patients have revealed a range of anomalies including polymicrogyria, absent or hypoplastic anterior commissure and absent or hypoplastic pineal gland (19, 41). The functional consequence of these changes is not yet known. The phenotypes were highly variable even in family members with the same mutation.
Three reports have documented behavioral anomalies in aniridia patients, including cognitive dysfunction (42), aggressive behavior (42, 43), autism (20), and mild mental retardation (43). Two of these cases were familial, with the behavioral phenotype segregating with aniridia (42, 43); however neither pedigree was large enough to prove linkage between the behavioral phenotype and the PAX6 mutation.
Four aniridia patients with PAX6 mutations all had glucose intolerance related to impaired insulin secretion (44). This is the first report of a possible link between PAX6 heterozygosity and impairment of pancreatic function.
LESSONS FROM THE MOUSE
Important insights into Pax6 function have come from the naturally occurring mouse Pax6 mutant small eye and from experimentally engineered animals. Many recent studies have highlighted the role of Pax6 in forebrain development including regionalization, cell migration, and axon guidance (40). The role of Pax6 in eye development has been covered in recent reviews (45, 46). Here I discuss the eye phenotype in heterozygous small eye mice and advances in our understanding of the regulation of Pax6 transcription.
Some Pax6 heterozygous mice have aniridia-like iris anomalies (2) but others have corneal opacities and lens-corneal adhesions that resemble Peters' anomaly (24, 46, 47;Fig. 3C). Detailed histologic studies of neonatal mice with Peters-like defects revealed that the lens frequently fails to separate from the cornea (48). Trabecular meshwork development was also abnormal in heterozygotes.
Cis-ACTING DNA ELEMENTS
The sequences responsible for regulating the spatially and temporally complex pattern of Pax6 transcription have come under intense scrutiny. Many of the key regulatory regions were initially identified in quail (49, 50) but have now been more rigorously analyzed in mice. The availability of genome sequence from different organisms has greatly facilitated the identification of potential regulatory elements, since regulatory sequences often show up as blocks of high nucleotide sequence conservation (50–56). The pattern of expression directed by each putative control element can be determined by reporter gene expression analysis in transgenic mice.
In mice, Pax6 expression can be initiated at two 5′ promoters, P0 and P1, both of which are associated with distinct regulatory elements (52, 53). P0 and P1 transcripts are both abundant in the lens placode but P0 transcripts predominate in the corneal and conjunctival epithelia while P1 transcripts predominate in the optic vesicle and CNS (52). Intron 4 contains a third potential promoter, Pα, and regulatory sequences that direct expression to the retina, ciliary body, and iris (52, 53, 57;Fig. 4).

The PAX6 locus, showing regulatory elements of the PAX6 gene and target genes of the PAX6 protein. (a) The PAX6 genomic region. The PAX6 gene is shown as a green rectangle. The last 3 exons of the neighboring ELP4 gene are shown as red rectangles (54, 61). The PAX6 protein is represented as a green oval. Examples of genes that are activated (+) or repressed (−) by the PAX6 protein are shown (63, 66, 68, 69, 72–78). Single arrows represent direct control; double arrows show that direct control has not yet been proven. (b) PAX6 regulatory elements. Regions of the PAX6 locus that contain control elements are shown in expanded form. Numbered green rectangles represent individual exons of the PAX6 gene. Lettered rectangles represent control elements that have been analyzed by reporter studies in transgenic mice. The expression patterns are as follows: A, endocrine pancreas (52, 53); B (the ectodermal element), surface ectoderm, lens, cornea (51, 53); C, telencephalon, hindbrain, spinal cord, photoreceptors (52, 53); D (the α-element), retina, iris, ciliary body, spinal cord (52, 53, 57); E, pretectum, neural retina, olfactory structures (55); F, lens, diencephalon, hindbrain (54); G, neural folds, optic primordia, optic vesicle, retina (54). Hypersensitive sites (HSS) that have been identified within the downstream regulatory region are indicated by arrows (54). Colored ovals represent proteins that directly bind and activate (SIX3, PAX6, SOX2, MEIS) or repress (PAX2) the regulatory elements (56, 66, 68, 69). Brackets indicate that PAX6 and SOX2 physically interact (69, 70).
A number of cis-acting regulatory elements lie far downstream of the 3′ end of the Pax6 gene. These were identified by studying chromosomal rearrangements in a small number of aniridia patients with breakpoints up to 130kb distal to the PAX6 coding region (58, 59). The downstream region was shown to be essential for normal eye development in transgenic mice (54, 60). Long-range sequence comparisons revealed several highly conserved noncoding elements in the downstream region that could direct expression to specific regions of the eye, brain, and olfactory system (54, 55;Fig. 4). When the mutant and normal chromosomes were separated in somatic cell hybrids, no Pax6 transcription could be detected from the allele on the rearranged chromosome, indicating that these downstream elements must be present in cis for normal Pax6 expression (59). Remarkably, the downstream elements are located in an intron of the neighboring ELP4 gene (61;Fig. 4). ELP4 encodes a subunit of an RNA polymerase-associated histone acetylase (62). Although the patients with chromosomal rearrangements necessarily lose one functional copy of ELP4, this appears to have no phenotypic consequence.
Physically distant elements may combine to bring about the correct expression pattern in any particular structure; lens expression is directed by two elements, one upstream of P0 (51, 53) and another over 150 kb away (54). Deletion of the upstream element reduces but does not abolish Pax6 expression in the lens placode suggesting that different elements may act in a combinatorial fashion to determine the correct level of transcription (63).
The well-studied β-globin gene cluster has given insights into the mechanism of long-range control of gene expression (64, 65). Recent evidence supports a model in which physically distant sites (such as a long-range enhancer and a promoter) interact directly, with the intervening chromatin looping out. This may create a chromatin domain that is permissive for transcription (64, 65).
Trans-ACTING FACTORS
Progress has also been made in identifying the proteins that bind to these regulatory elements. The Pax6 α-element (box D, Fig. 4) contains two sites that bind both Pax2 and Pax6 (66). Pax2 represses, while Pax6 activates, reporter expression from this element. Competition between Pax6 and Pax2 binding may help to define the sharp boundary of Pax6 and Pax2 expression between the optic cup and the optic stalk (66).
The ectodermal element (box B, Fig. 4) binds the homeoproteins Six3, Meis1, and Meis2, which are expressed in the lens placode (56, 67, 68). Pax6 expression is up regulated in the lenses of transgenic mice that over-express Meis2 (56).
The ectodermal element also binds the −5a isoform of the Pax6 protein (which lacks the additional 14 amino acids in the paired domain), and the Sox2 and Sox3 proteins (69). Both Sox2 and Sox3 can synergistically activate the enhancer when co-expressed with Pax6, and Sox2 and Pax6 physically interact as judged by a co-immunoprecipitation assay with tagged proteins (69). Pax6 and Sox2 also form a co-DNA binding complex on the chick δ-crystallin enhancer (70). SOX2 gene mutations were recently described in anophthalmia patients (71). Sox2 may be an important co-factor of Pax6 during many key stages of eye development.
The Pax6, Six3, Meis, and Sox binding sites lie within 60 bp of each other in the core of the ectodermal enhancer (53, 56, 68, 69). This high density of sites hints at how Pax6 transcription can be fine-tuned by specific combinations of trans-acting factors in different cell types.
DOWNSTREAM TARGETS OF THE Pax6 PROTEIN
Genes that are directly activated by the Pax6 protein include Pax6 itself (66, 69), bHLH genes such as Ngn2(72) and Maf(73), homeobox genes such as Six3(68), crystallin genes (74, 75) and pancreatic hormone genes such as glucagon (76;Fig. 4). Pax6 is a repressor of Pax2 and β-crystallin(66, 77). Expression of the calcium-binding protein gene Necab and the forkhead gene Foxe3 are both absent in mutant mice, suggesting that they are downstream of Pax6, but a direct interaction has not yet been demonstrated (63, 78;Fig. 4).
For most developmental genes associated with human disease, a handful of mutations are described before attention turns to functional studies in animal models. In the case of PAX6 however, large numbers of mutations are still being reported, thus providing a rich molecular pathology backed up by extensive functional work. The challenge ahead is to better understand the phenotypes associated with different mutations in terms of functional information gained from model systems.
Abbreviations
- kb:
-
kilobases
- MRI:
-
magnetic resonance imaging
- PST:
-
proline, serine, and threonine-rich domain
- WAGR:
-
Wilms tumor, aniridia, genitourinary malformations and mental retardation
REFERENCES
Ton CCT, Hirvonen H, Miwa H, Weil MM, Monaghan P, Jordan T, van Heyningen V, Hastie ND, Meijers-Heijboer H, Drechsler M, Royer-Pokora B, Collins F, Swaroop A, Strong LC, Saunders GF 1991 Positional cloning and characterization of a paired-box and homeobox-containing gene from the aniridia region. Cell 67: 1059–1074
Jordan T, Hanson I, Zaletayev D, Hodgson S, Prosser J, Seawright A, Hastie N, van Heyningen V 1992 The human PAX6 gene is mutated in two patients with aniridia. Nat Genet 1: 328–332
Glaser T, Walton DS, Maas RL 1992 Genomic structure, evolutionary conservation and aniridia mutations in the human PAX6 gene. Nat Genet 2: 232–239
Hanson IM, Seawright A, Hardman K, Hodgson S, Zaletayev D, Fekete G, van Heyningen V 1993 PAX6 mutations in aniridia. Hum Mol Genet 2: 915–920
Martha A, Ferrell RE, Mintz-Hittner H, Lyons LA, Saunders GF 1994 Paired box mutations in familial and sporadic aniridia predicts truncated aniridia proteins. Am J Hum Genet 54: 801–811
Hill RE, Favor J, Hogan BL, Ton CC, Saunders GF, Hanson IM, Prosser J, Jordan T, Hastie ND, van Heyningen V 1991 Mouse small eye results from mutations in a paired-like homeobox-containing gene. Nature 354: 522–525
Wilson DS, Guenther B, Desplan C, Kuriyan J 1995 High resolution crystal structure of a paired (Pax) class cooperative homeodomain dimer on DNA. Cell 82: 709–719
Xu HE, Rould MA, Xu W, Epstein JA, Maas RL, Pabo CO 1999 Crystal structure of the human Pax6 paired domain-DNA complex reveals specific roles for the linker region and carboxy-terminal subdomain in DNA binding. Genes Dev 13: 1263–1275
Glaser T, Jepeal L, Edwards JG, Young SR, Favor J, Maas RL 1994 PAX6 gene dosage effect in a family with congenital cataracts, aniridia, anophthalmia and central nervous system defects. Nat Genet 7: 463–471
Singh S, Chao LY, Mishra R, Davies J, Saunders GF 2001 Missense mutation at the C-terminus of PAX6 negatively modulates homeodomain function. Hum Mol Genet 10: 911–918
Crolla JA, Van Heyningen V 2002 Frequent chromosome aberrations revealed by molecular cytogenetic studies in patients with aniridia. Am J Hum Genet 71: 1138–1149
Prosser J, van Heyningen V 1998 PAX6 mutations reviewed. Hum Mutat 11: 93–108
Brown A, McKie M, van Heyningen V, Prosser J 1998 The human PAX6 mutation database. Nucleic Acids Res 26: 259–264
Culbertson MR 1999 RNA surveillance: unforeseen consequences for gene expression, inherited genetic disorders and cancer. Trends Genet 15: 74–80
Byers PH 2002 Killing the messenger: new insights into nonsense-mediated mRNA decay. J Clin Invest 109: 3–6
Vincent MC, Pujo AL, Olivier D, Calvas P 2003 Screening for PAX6 gene mutations is consistent with haploinsufficiency as the main mechanism leading to various ocular defects. Eur J Hum Genet 11: 163–169
Baum L, Pang CP, Fan DS, Poon PM, Leung YF, Chua JK, Lam DS 1999 Run-on mutation and three novel nonsense mutations identified in the PAX6 gene in patients with aniridia. Hum Mutat 14: 272–273
Gronskov K, Olsen JH, Sand A, Pedersen W, Carlsen N, Bak Jylling AM, Lyngbye T, Brondum-Nielsen K, Rosenberg T 2001 Population-based risk estimates of Wilms tumor in sporadic aniridia. A comprehensive mutation screening procedure of PAX6 identifies 80% of mutations in aniridia. Hum Genet 109: 11–18
Sisodiya SM, Free SL, Williamson KA, Mitchell TN, Willis C, Stevens JM, Kendall BE, Shorvon SD, Hanson IM, Moore AT, van Heyningen V 2001 PAX6 haploinsufficiency causes cerebral malformation and olfactory dysfunction in humans. Nat Genet 28: 214–216
Chao LY, Mishra R, Strong LC, Saunders GF 2003 Missense mutations in the DNA-binding region and termination codon in PAX6. Hum Mutat 21: 138–145
Tang HK, Chao LY, Saunders GF 1997 Functional analysis of paired box missense mutations in the PAX6 gene. Hum Mol Genet 6: 381–386
Gronskov K, Rosenberg T, Sand A, Brondum-Nielsen K 1999 Mutational analysis of PAX6: 16 novel mutations including 5 missense mutations with a mild aniridia phenotype. Eur J Hum Genet 7: 274–286
Hanson I, Churchill A, Love J, Axton R, Moore T, Clarke M, Meire F, van Heyningen V 1999 Missense mutations in the most ancient residues of the PAX6 paired domain underlie a spectrum of human congenital eye malformations. Hum Mol Genet 8: 165–172
Hanson IM, Fletcher JM, Jordan T, Brown A, Taylor D, Adams RJ, Punnett HH, van Heyningen V 1994 Mutations at the PAX6 locus are found in heterogeneous anterior segment malformations including Peters' anomaly. Nat Genet 6: 168–173
Azuma N, Nishina S, Yanagisawa H, Okuyama T, Yamada M 1996 PAX6 missense mutation in isolated foveal hypoplasia. Nat Genet 13: 141–142
Azuma N, Yamada M 1998 Missense mutation at the C terminus of the PAX6 gene in ocular anterior segment anomalies. Invest Ophthalmol Vis Sci 39: 828–830
Azuma N, Yamaguchi Y, Handa H, Hayakawa M, Kanai A, Yamada M 1999 Missense mutation in the alternative splice region of the PAX6 gene in eye anomalies. Am J Hum Genet 65: 656–663
van Heyningen V, Williamson KA 2002 PAX6 in sensory development. Hum Mol Genet 11: 1161–1167
Azuma N, Yamaguchi Y, Handa H, Tadokoro K, Asaka A, Kawase E, Yamada M 2003 Mutations of the PAX6 gene detected in patients with a variety of optic-nerve malformations. Am J Hum Genet 72: 1565–1570
Morrison D, FitzPatrick D, Hanson I, Williamson K, van Heyningen V, Fleck B, Jones I, Chalmers J, Campbell H 2002 National study of microphthalmia, anophthalmia, and coloboma (MAC) in Scotland: investigation of genetic aetiology. J Med Genet 39: 16–22
Epstein JA, Glaser T, Cai J, Jepeal L, Walton DS, Maas RL 1994 Two independent and interactive DNA-binding subdomains of the Pax6 paired domain are regulated by alternative splicing. Genes Dev 8: 2022–2034
Singh S, Mishra R, Arango NA, Deng JM, Behringer RR, Saunders GF 2002 Iris hypoplasia in mice that lack the alternatively spliced Pax6(5a) isoform. Proc Natl Acad Sci USA 99: 6812–6815
Dome JS, Coppes MJ 2002 Recent advances in Wilms tumor genetics. Curr Opin Pediatr 14: 5–11
Drechsler M, Meijers-Heijboer EJ, Schneider S, Schurich B, Grond-Ginsbach C, Tariverdian G, Kantner G, Blankenagel A, Kaps D, Schroeder-Kurth T, Royer-Pokora B 1994 Molecular analysis of aniridia patients for deletions involving the Wilms' tumor gene. Hum Genet 94: 331–338
Muto R, Yamamori S, Ohashi H, Osawa M 2002 Prediction by FISH analysis of the occurrence of Wilms tumor in aniridia patients. Am J Med Genet 108: 285–289
Fantes JA, Bickmore WA, Fletcher JM, Ballesta F, Hanson IM, van Heyningen V 1992 Submicroscopic deletions at the WAGR locus, revealed by nonradioactive in situ hybridization. Am J Hum Genet 51: 1286–1294
Kawase E, Tanaka K, Honna T, Azuma N 2001 A case of atypical WAGR syndrome with anterior segment anomaly and microphthalmos. Arch Ophthalmol 119: 1855–1856
Riise R, Storhaug K, Brondum-Nielsen K 2001 Rieger syndrome is associated with PAX6 deletion. Acta Ophthalmol Scand 79: 201–203
Alward WL 2000 Axenfeld-Rieger syndrome in the age of molecular genetics. Am J Ophthalmol 130: 107–115
Simpson TI, Price DJ 2002 Pax6; a pleiotropic player in development. Bioessays 24: 1041–1051
Mitchell TN, Free SL, Williamson KA, Stevens JM, Churchill AJ, Hanson IM, Shorvon SD, Moore AT, Van Heyningen V, Sisodiya SM 2003 Polymicrogyria and absence of pineal gland due to PAX6 mutation. Ann Neurol 53: 658–663
Heyman I, Frampton I, van Heyningen V, Hanson I, Teague P, Taylor A, Simonoff E 1999 Psychiatric disorder and cognitive function in a family with an inherited novel mutation of the developmental control gene PAX6. Psychiatr Genet 9: 85–90
Malandrini A, Mari F, Palmeri S, Gambelli S, Berti G, Bruttini M, Bardelli AM, Williamson K, van Heyningen V, Renieri A 2001 PAX6 mutation in a family with aniridia, congenital ptosis, and mental retardation. Clin Genet 60: 151–154
Yasuda T, Kajimoto Y, Fujitani Y, Watada H, Yamamoto S, Watarai T, Umayahara Y, Matsuhisa M, Gorogawa S, Kuwayama Y, Tano Y, Yamasaki Y, Hori M 2002 PAX6 mutation as a genetic factor common to aniridia and glucose intolerance. Diabetes 51: 224–230
Ashery-Padan R, Gruss P 2001 Pax6 lights-up the way for eye development. Curr Opin Cell Biol 13: 706–714
Pichaud F, Desplan C 2002 Pax genes and eye organogenesis. Curr Opin Genet Dev 12: 430–434
Thaung C, West K, Clark BJ, McKie L, Morgan JE, Arnold K, Nolan PM, Peters J, Hunter AJ, Brown SD, Jackson IJ, Cross SH 2002 Novel ENU-induced eye mutations in the mouse: models for human eye disease. Hum Mol Genet 11: 755–767
Baulmann DC, Ohlmann A, Flugel-Koch C, Goswami S, Cvekl A, Tamm ER 2002 Pax6 heterozygous eyes show defects in chamber angle differentiation that are associated with a wide spectrum of other anterior eye segment abnormalities. Mech Dev 118: 3–17
Plaza S, Dozier C, Turque N, Saule S 1995 Quail Pax-6 (Pax-QNR) mRNAs are expressed from two promoters used differentially during retina development and neuronal differentiation. Mol Cell Biol 15: 3344–3353
Plaza S, Dozier C, Langlois MC, Saule S 1995 Identification and characterization of a neuroretina-specific enhancer element in the quail Pax-6 (Pax-QNR) gene. Mol Cell Biol 15: 892–903
Williams SC, Altmann CR, Chow RL, Hemmati-Brivanlou A, Lang RA 1998 A highly conserved lens transcriptional control element from the Pax-6 gene. Mech Dev 73: 225–229
Xu PX, Zhang X, Heaney S, Yoon A, Michelson AM, Maas RL 1999 Regulation of Pax6 expression is conserved between mice and flies. Development 26: 383–395
Kammandel B, Chowdhury K, Stoykova A, Aparicio S, Brenner S, Gruss P 1999 Distinct cis-essential modules direct the time-space pattern of the Pax6 gene activity. Dev Biol 205: 79–97
Kleinjan DA, Seawright A, Schedl A, Quinlan RA, Danes S, van Heyningen V 2001 Aniridia-associated translocations, DNase hypersensitivity, sequence comparison and transgenic analysis redefine the functional domain of PAX6. Hum Mol Genet 10: 2049–2059
Griffin C, Kleinjan DA, Doe B, van Heyningen V 2002 New 3′ elements control Pax6 expression in the developing pretectum, neural retina and olfactory region. Mech Dev 112: 89–100
Zhang X, Friedman A, Heaney S, Purcell P, Maas RL 2002 Meis homeoproteins directly regulate Pax6 during vertebrate lens morphogenesis. Genes Dev 16: 2097–2107
Xu ZP, Saunders GF 1998 PAX6 intronic sequence targets expression to the spinal cord. Dev Genet 23: 259–263
Fantes J, Redeker B, Breen M, Boyle S, Brown J, Fletcher J, Jones S, Bickmore W, Fukushima Y, Mannens M, Danes S, van Heyningen V, Hanson I 1995 Aniridia-associated cytogenetic rearrangements suggest that a position effect may cause the mutant phenotype. Hum Mol Genet 4: 415–422
Lauderdale JD, Wilensky JS, Oliver ER, Walton DS, Glaser T 2000 3′ deletions cause aniridia by preventing PAX6 gene expression. Proc Natl Acad Sci 97: 13755–13759
Schedl A, Ross A, Lee M, Engelkamp D, Rashbass P, van Heyningen V, Hastie ND 1996 Influence of PAX6 gene dosage on development: overexpression causes severe eye abnormalities. Cell 86: 71–82
Kleinjan DA, Seawright A, Elgar G, van Heyningen V 2002 Characterization of a novel gene adjacent to PAX6, revealing synteny conservation with functional significance. Mamm Genome 13: 102–107
Winkler GS, Petrakis TG, Ethelberg S, Tokunaga M, Erdjument-Bromage H, Tempst P, Svejstrup JQ 2001 RNA polymerase II elongator holoenzyme is composed of two discrete subcomplexes. J Biol Chem 276: 32743–32749
Dimanlig PV, Faber SC, Auerbach W, Makarenkova HP, Lang RA 2001 The upstream ectoderm enhancer in Pax6 has an important role in lens induction. Development 128: 4415–4424
Tolhuis B, Palstra RJ, Splinter E, Grosveld F, de Laat W 2002 Looping and interaction between hypersensitive sites in the active beta-globin locus. Mol Cell 10: 1453–1465
Carter D, Chakalova L, Osborne CS, Dai YF, Fraser P 2002 Long-range chromatin regulatory interactions in vivo. Nat Genet 32: 623–626
Schwarz M, Cecconi F, Bernier G, Andrejewski N, Kammandel B, Wagner M, Gruss P 2000 Spatial specification of mammalian eye territories by reciprocal transcriptional repression of Pax2 and Pax6. Development 127: 4325–4334
Jean D, Bernier G, Gruss P 1999 Six6 (Optx2) is a novel murine Six3-related homeobox gene that demarcates the presumptive pituitary/hypothalamic axis and the ventral optic stalk. Mech Dev 84: 31–40
Goudreau G, Petrou P, Reneker LW, Graw J, Loster J, Gruss P 2002 Mutually regulated expression of Pax6 and Six3 and its implications for the Pax6 haploinsufficient lens phenotype. Proc Natl Acad Sci USA 99: 8719–8724
Aota S, Nakajima N, Sakamoto R, Watanabe S, Ibaraki N, Okazaki K 2003 Pax6 autoregulation mediated by direct interaction of Pax6 protein with the head surface ectoderm-specific enhancer of the mouse Pax6 gene. Dev Biol 257: 1–13
Kamachi Y, Uchikawa M, Tanouchi A, Sekido R, Kondoh H 2001 Pax6 and SOX2 form a co-DNA-binding partner complex that regulates initiation of lens development. Genes Dev 15: 1272–1286
Fantes J, Ragge NK, Lynch SA, McGill NI, Collin JR, Howard-Peebles PN, Hayward C, Vivian AJ, Williamson K, Van Heyningen V, FitzPatrick DR 2003 Mutations in SOX2 cause anophthalmia. Nat Genet 33: 461–463
Marquardt T, Ashery-Padan R, Andrejewski N, Scardigli R, Guillemot F, Gruss P 2001 Pax6 is required for the multipotent state of retinal progenitor cells. Cell 105: 43–55
Sakai M, Serria MS, Ikeda H, Yoshida K, Imaki J, Nishi S 2001 Regulation of c-maf gene expression by Pax6 in cultured cells. Nucleic Acids Res 29: 1228–1237
Cvekl A, Kashanchi F, Sax CM, Brady JN, Piatigorsky J 1995 Transcriptional regulation of the mouse alpha A-crystallin gene: activation dependent on a cyclic AMP-responsive element (DE1/CRE) and a Pax-6-binding site. Mol Cell Biol 15: 653–660
Richardson J, Cvekl A, Wistow G 1995 Pax-6 is essential for lens-specific expression of zeta-crystallin. Proc Natl Acad Sci USA 92: 4676–4680
Planque N, Leconte L, Coquelle FM, Benkhelifa S, Martin P, Felder-Schmittbuhl MP, Saule S 2001 Interaction of Maf transcription factors with Pax-6 results in synergistic activation of the glucagon promoter. J Biol Chem 276: 35751–35760
Duncan MK, Haynes JI, Cvekl A, Piatigorsky J 1998 Dual roles for Pax-6: a transcriptional repressor of lens fiber cell-specific beta-crystallin genes. Mol Cell Biol 18: 5579–5586
Bernier G, Vukovich W, Neidhardt L, Herrmann BG, Gruss P 2001 Isolation and characterization of a downstream target of Pax6 in the mammalian retinal primordium. Development 128: 3987–3994
Author information
Authors and Affiliations
Corresponding author
Additional information
This work was supported in the form of a Career Development Award from the UK Medical Research Council.
This review is the ninth in this series. Dr. Hanson describes the remarkable studies that have determined the relationship between the PAX6 gene and eye development. These studies include observations of anomalies in humans and also experimental studies in animals to determine the mechanism of action of the gene. In children with aniridia, the relationship between this determinant of ocular development and the WT1 gene of Wilms Tumor is discussed. The PAX6 gene is a prime example of a specific gene responsible for normal human development.
Alvin Zipursky
Editor-in-Chief
Rights and permissions
About this article
Cite this article
Hanson, I. PAX6 and Congenital Eye Malformations. Pediatr Res 54, 791–796 (2003). https://doi.org/10.1203/01.PDR.0000096455.00657.98
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/01.PDR.0000096455.00657.98
This article is cited by
-
A novel PAX6 variant as the cause of aniridia in a Chinese patient with SRRRD
BMC Medical Genomics (2023)
-
Whole-genome sequencing of multiple related individuals with type 2 diabetes reveals an atypical likely pathogenic mutation in the PAX6 gene
European Journal of Human Genetics (2023)
-
Diagnostik, Klinik und Genetik kongenitaler Hornhauttrübungen
Der Ophthalmologe (2022)
-
TRPM3_miR-204: a complex locus for eye development and disease
Human Genomics (2020)
-
Autosomal dominant foveal hypoplasia without visible macular abnormalities and PAX6 mutations
Japanese Journal of Ophthalmology (2020)
